UPS vs. Autophagy: Mechanisms, Applications, and Therapeutic Targeting in Misfolded Protein Degradation
This article provides a comprehensive comparison of the Ubiquitin-Proteasome System (UPS) and Autophagy, the two primary cellular pathways for degrading misfolded proteins.
UPS vs. Autophagy: Mechanisms, Applications, and Therapeutic Targeting in Misfolded Protein Degradation
Abstract
This article provides a comprehensive comparison of the Ubiquitin-Proteasome System (UPS) and Autophagy, the two primary cellular pathways for degrading misfolded proteins. Targeting researchers and drug development professionals, we explore the foundational mechanisms of each system, from ubiquitin tagging and proteasomal hydrolysis to autophagosome formation and lysosomal degradation. The review delves into advanced methodological applications, including proteolysis-targeting chimeras (PROTACs) and autophagy-targeting chimeras (AUTACs), and analyzes the crosstalk and compensatory dynamics between these pathways. We further address troubleshooting in disease contexts like neurodegeneration and cancer, where these systems are impaired, and offer a comparative validation of their distinct substrate scopes, efficiency, and therapeutic potential. The synthesis aims to inform the strategic selection and optimization of targeted protein degradation technologies for novel therapeutics.
Core Mechanisms: Deconstructing the UPS and Autophagy Pathways
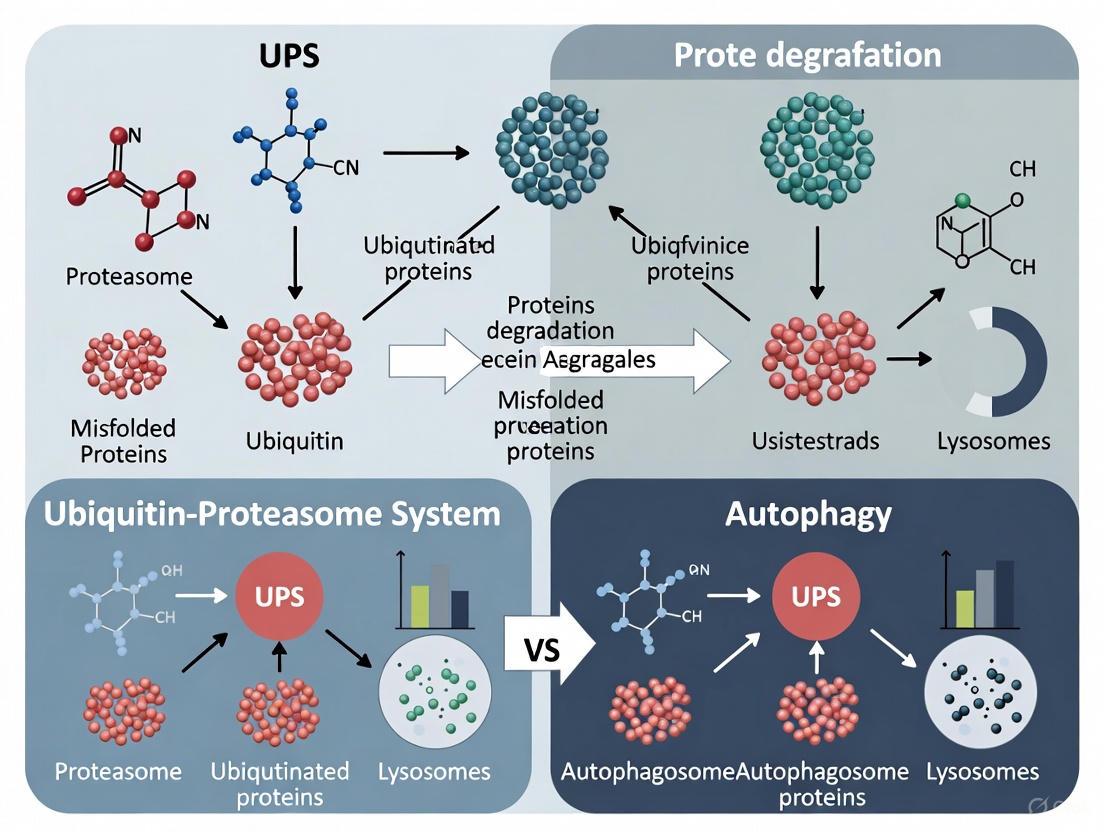
In eukaryotic cells, the ubiquitin-proteasome system (UPS) and autophagy represent the two major pathways responsible for intracellular protein degradation and recycling. While both systems are essential for maintaining cellular homeostasis, they differ fundamentally in their mechanisms, substrate preferences, and biological roles [1] [2]. The UPS functions as a highly selective degradation machinery primarily targeting short-lived regulatory proteins and soluble misfolded proteins for rapid destruction [3] [2]. In contrast, autophagy specializes in the bulk degradation of long-lived proteins, insoluble protein aggregates, and damaged organelles through the lysosomal pathway [4] [2]. This comparative analysis examines the mechanistic operation, substrate specificity, and experimental approaches for studying the UPS, with particular emphasis on its role in degrading misfolded proteins relative to autophagic pathways.
The UPS and autophagy employ distinct molecular machinery and operate through different cellular mechanisms. The UPS is characterized by its high specificity for individual proteins and rapid degradation kinetics, while autophagy handles larger cellular components through vesicular trafficking [1] [5].
Table 1: Fundamental Characteristics of UPS and Autophagy
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy |
|---|---|---|
| Primary Function | Selective degradation of short-lived proteins | Bulk degradation of long-lived proteins and organelles |
| Degradation Site | Proteasome (cytosol/nucleus) | Lysosome (autolysosome) |
| Mechanism | ATP-dependent proteolysis | Vesicular trafficking & lysosomal hydrolysis |
| Key Molecular Tags | K48-linked polyubiquitin chains | K63-linked ubiquitin chains, LC3-binding |
| Temporal Regulation | Rapid (minutes) | Slower (hours) |
| Energy Requirements | ATP for ubiquitination & proteasomal degradation | ATP for autophagosome formation & trafficking |
| Selectivity Mediators | E3 ubiquitin ligases, ubiquitin receptors | Autophagy receptors (p62, NBR1, HDAC6) |
The UPS is responsible for degrading approximately 80-90% of cellular proteins, including many regulatory proteins that require temporal control, such as cell cycle regulators and transcription factors [3]. Autophagy, while capable of selective degradation, primarily functions as a stress response system that scavenges nutrients during starvation and eliminates damaged organelles [1] [4].
Mechanistic Operation of the UPS
The Ubiquitination Cascade
Protein degradation via the UPS begins with a precise ubiquitination process involving three enzyme classes [3] [2] [5]:
- E1 Ubiquitin-Activating Enzymes: Initiate the pathway by activating ubiquitin in an ATP-dependent manner. Humans possess only two E1 enzymes, creating an initial bottleneck in the system [3] [5].
- E2 Ubiquitin-Conjugating Enzymes: Carry the activated ubiquitin to the target substrate. With approximately 37 E2 enzymes in humans, this step provides moderate specificity [3] [5].
- E3 Ubiquitin Ligases: Confer substrate specificity by recognizing target proteins and facilitating ubiquitin transfer. With over 600 E3 ligases in humans, this step provides the greatest diversity and specificity in the system [2] [5].
The ubiquitination process creates polyubiquitin chains linked through lysine 48 (K48) of ubiquitin, which serves as the primary degradation signal for the proteasome [3] [2]. A chain of four or more ubiquitin molecules is generally necessary and sufficient for proteasomal recognition [1].
The Proteasome Complex
The 26S proteasome is a massive 2.5 MDa proteolytic complex consisting of two primary components [1] [2]:
- 20S Core Particle: Barrel-shaped structure containing three distinct proteolytic activities (caspase-like, trypsin-like, and chymotrypsin-like) in its inner chamber [2].
- 19S Regulatory Particle: Recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds target proteins, and translocates them into the 20S core for degradation [1] [2].
The proteasome degrades target proteins into small peptides (3-25 amino acids), which are further processed to amino acids by cellular peptidases and recycled for new protein synthesis [2].
Diagram 1: UPS mechanism: ubiquitin tagging and proteasome degradation.
Comparative Degradation of Misfolded Proteins
Substrate Specificity and Selection Mechanisms
The UPS and autophagy employ different strategies for recognizing and processing misfolded proteins, with the nature of the misfolded protein determining its degradation pathway [1] [6].
Table 2: Misfolded Protein Degradation Pathways
| Misfolded Protein Characteristic | Primary Degradation Pathway | Recognition Mechanism | Experimental Evidence |
|---|---|---|---|
| Soluble cytosolic proteins | UPS | CHIP E3 ligase with Hsp70/Hsp40 chaperones | CHIP knockdown impairs degradation of cytosolic misfolded proteins [6] |
| ER luminal proteins | ERAD (UPS-dependent) | HRD1 E3 ligase complex with ER chaperones (BiP, calnexin) | HRD1 inhibition blocks degradation of ER luminal substrates [6] |
| Aggregation-prone proteins | Autophagy | p62/SQSTM1 links ubiquitin to LC3 on autophagosomes | p62 deletion impairs aggregate clearance [1] |
| Damaged organelles | Selective autophagy | Specific receptors (e.g., mitophagy receptors) | Receptor mutations prevent organelle turnover [2] |
| Membrane proteins with cytosolic lesions | UPS (via ERAD) | RMA1 E3 ligase with DNAJB12 chaperone | RMA1/DNAJB12 complex recognizes CFTR folding defects [6] |
The ubiquitin code plays a crucial role in directing substrates to the appropriate pathway. K48-linked polyubiquitin chains typically target proteins for proteasomal degradation, while K63-linked chains and monoubiquitination often signal for autophagic clearance [1] [2]. Autophagy adaptor proteins such as p62, NBR1, and HDAC6 contain both ubiquitin-binding domains and LC3-interacting regions, enabling them to bridge ubiquitinated cargo to the autophagic machinery [1].
Endoplasmic Reticulum-Associated Degradation (ERAD)
ERAD represents a specialized UPS pathway that eliminates misfolded proteins from the endoplasmic reticulum [7] [6]. This process involves:
- Recognition: Misfolded ER proteins are identified by chaperones and lectins that detect folding defects [7].
- Retrotranslocation: Recognized substrates are extracted from the ER membrane via the p97/VCP ATPase complex in an ATP-dependent process [7] [6].
- Ubiquitination: ER-associated E3 ligases (HRD1, GP78, RMA1) conjugate ubiquitin chains to target proteins [6].
- Degradation: Ubiquitinated substrates are delivered to proteasomes for destruction [7].
When ERAD is impaired, cells experience reduced ER stress and activate alternative degradation pathways, including lysosomal degradation, demonstrating the crosstalk between these systems [8].
Experimental Approaches and Methodologies
Investigating UPS Function and Inhibition
Studying UPS activity requires specific methodological approaches that can distinguish its function from autophagy and other degradation pathways:
Proteasome Inhibition Assays
- Pharmacological Inhibition: Use specific proteasome inhibitors (e.g., MG132, bortezomib, lactacystin) at appropriate concentrations (typically 1-10 μM) for defined time periods (4-24 hours) to measure substrate accumulation [9].
- Genetic Approaches: siRNA or CRISPR-mediated knockdown of specific proteasome subunits or E3 ligases to assess their role in substrate turnover.
- Reporter Substrates: Express ubiquitin fusion degradation (UFD) substrates such as Ub-G76V-GFP or other well-characterized UPS reporters to monitor proteasome activity in live cells.
UPS Activity Measurements
- In Vitro Proteasome Activity Assays: Use fluorogenic substrates (e.g., Suc-LLVY-AMC for chymotrypsin-like activity) in cell lysates to directly measure proteasome function.
- Ubiquitin Chain Accumulation: Monitor polyubiquitinated protein levels by western blotting following proteasome inhibition.
- Pulse-Chase Analysis: Track degradation kinetics of specific proteins using radioactive or stable isotope labeling.
Experimental Considerations
- Simultaneous Autophagy Inhibition: When specifically studying UPS, consider using autophagy inhibitors (e.g., chloroquine, bafilomycin A1) to prevent compensatory autophagy activation [4].
- Time Course Analysis: Perform experiments at multiple time points, as acute versus chronic proteasome inhibition engages different compensatory mechanisms [9].
- Stress Induction: Use ER stress inducers (tunicamycin, thapsigargin) or proteotoxic agents to study UPS function under stress conditions [7].
Model Substrates for Misfolded Protein Degradation
Several well-characterized model substrates have been developed to study misfolded protein degradation:
SZ* Substrate System The SZ* model substrate incorporates a truncated cytosolic nucleotide-binding domain (NBD2*) as a degron fused to a single transmembrane domain [7]. This construct allows researchers to:
- Monitor partitioning between ERAD and post-ER quality control pathways
- Assess the impact of aggregation propensity on degradation route selection
- Manipulate experimental conditions (heat shock, overexpression) to shift degradation preferences [7]
Key Finding: Substrates with higher aggregation propensity are preferentially retained in the ER and targeted for ERAD rather than post-ER quality control mechanisms [7].
Research Reagent Solutions
Table 3: Essential Reagents for Studying UPS and Protein Degradation
| Reagent Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib, Lactacystin | Reversible or irreversible proteasome inhibition | Concentration and time-dependent effects; monitor compensatory autophagy [9] |
| E1 Inhibitors | TAK-243, PYR-41 | Block ubiquitin activation | Broad UPS inhibition; high toxicity |
| E3 Ligase Modulators | MLN4924 (NEDD8-activating enzyme inhibitor) | Indirect E3 ligase regulation | Affects cullin-RING ligases specifically |
| UPS Reporters | Ub-G76V-GFP, Ub-R-GFP | Fluorescent UPS substrate reporters | Accumulate when UPS impaired; quantifiable by flow cytometry or microscopy |
| ER Stress Inducers | Tunicamycin, Thapsigargin | Induce ER protein misfolding | Activate ERAD pathway; monitor UPR induction |
| p97/VCP Inhibitors | CB-5083, DBeQ | Block ERAD substrate extraction | Impair ERAD specifically; validate ERAD substrates |
| Autophagy Inhibitors | Chloroquine, Bafilomycin A1, 3-MA | Block autophagic degradation | Distinguish UPS vs. autophagy contributions |
| Ubiquitin Binding Reagents | TUBE (Tandem Ubiquitin Binding Entities) | Enrich polyubiquitinated proteins | Isolate UPS substrates from complex mixtures |
Pathway Crosstalk and Compensatory Mechanisms
Growing evidence reveals extensive crosstalk and coordination between the UPS and autophagy, particularly in response to proteotoxic stress [1] [4] [2]. Key integration points include:
Molecular Interfaces
Several molecules function at the interface between UPS and autophagy:
- Ubiquitin: Serves as a common degradation signal for both pathways, with chain topology determining route specificity [1] [2].
- p62/SQSTM1: Autophagy adaptor that recognizes ubiquitinated proteins and links them to LC3 on autophagosomes; accumulates when autophagy is impaired [1].
- HDAC6: Cytoplasmic deacetylase that recognizes ubiquitinated proteins and facilitates their transport to aggresomes for autophagic clearance [1].
- CHIP E3 Ubiquitin Ligase: Functions in both pathways, directing some substrates to proteasomes and others to autophagy through interactions with different co-chaperones [1] [6].
Compensatory Activation
When one degradation system is impaired, the other often compensates:
- Proteasome Inhibition: Leads to increased autophagy activation through multiple mechanisms, including accumulation of ubiquitinated proteins and activation of stress response pathways [4] [2].
- Autophagy Inhibition: Can enhance UPS activity, though this compensatory relationship is less robust than the reverse [4].
- Transcription Factor Coordination: Transcription factors like NRF1 activate proteasome subunit expression when proteasome capacity is insufficient, while TFEB coordinates lysosomal and autophagic gene expression [9].
Diagram 2: Cellular protein degradation crosstalk between UPS and autophagy.
The ubiquitin-proteasome system represents a sophisticated, highly selective mechanism for protein turnover that operates in continuous dialogue with autophagic pathways to maintain proteostasis. Its precision in targeting specific proteins for degradation, rapid kinetics, and central role in regulatory processes distinguishes it from the bulk degradation capabilities of autophagy. Understanding the specialized functions, substrate preferences, and compensatory relationships between these pathways provides crucial insights for developing targeted therapeutic interventions for protein aggregation diseases, cancer, and neurodegenerative disorders where proteostasis is compromised. The experimental frameworks and reagents outlined here provide essential tools for researchers dissecting the contributions of these systems to cellular health and disease.
The autophagy-lysosome pathway (ALP) represents a crucial intracellular degradation system, evolving from its initial characterization as a non-selective bulk degradation process to a highly sophisticated system for specific cargo recognition. This review systematically compares the ALP with the ubiquitin-proteasome system (UPS), examining their distinct yet complementary roles in cellular proteostasis. We provide detailed analysis of their mechanisms, substrate preferences, and functional specializations, supported by experimental data and methodological protocols. The emerging paradigm reveals that selective autophagy employs specific cargo receptors that recognize ubiquitinated substrates, creating a sophisticated interface between UPS and ALP. Understanding these intricate degradation pathways provides critical insights for developing targeted therapeutic interventions for neurodegenerative diseases, cancer, and other conditions characterized by proteostasis dysfunction.
Eukaryotic cells maintain protein homeostasis through two primary degradation systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP). While both systems utilize ubiquitin as a degradation signal, they differ fundamentally in their mechanisms, capacity, and substrate specificity [10]. The UPS primarily degrades short-lived soluble proteins through the 26S proteasome in an ATP-dependent process, requiring substrate unfolding [11] [10]. In contrast, the ALP degrades long-lived proteins, protein aggregates, damaged organelles, and intracellular pathogens through lysosomal hydrolases, capable of processing large macromolecular complexes and organelles without requiring complete unfolding [11] [10] [12].
The traditional view delineated these systems as operating independently—UPS handling targeted protein degradation and ALP managing bulk cytoplasmic clearance. However, emerging research reveals extensive crosstalk, particularly through ubiquitin signaling, which serves as a common degradation signal for both pathways [10]. This review systematically compares these systems, focusing on the sophisticated cargo recognition mechanisms that underlie selective autophagy and its relationship with UPS-mediated degradation.
Comparative Analysis: UPS vs. Autophagy-Lysosome Pathway
Table 1: Fundamental Characteristics of UPS and Autophagy-Lysosome Pathway
| Characteristic | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome Pathway (ALP) |
|---|---|---|
| Primary substrates | Short-lived proteins, misfolded soluble proteins [10] | Long-lived proteins, protein aggregates, damaged organelles, intracellular pathogens [11] [10] [12] |
| Degradation mechanism | ATP-dependent proteolysis via 26S proteasome complex [10] | Acid hydrolases within lysosomes [12] |
| Ubiquitin linkage preference | Primarily K48-linked chains [13] | K63-linked, K27-linked chains [13] |
| Membrane involvement | Not membrane-bound | Involves membrane dynamics (phagophore, autophagosome, lysosome) [11] |
| Degradation capacity | Limited to individual proteins | Can degrade large protein aggregates and entire organelles [10] [12] |
| Energy requirements | ATP-dependent for ubiquitination and proteasomal degradation [10] | ATP-dependent for membrane formation and fusion events [11] |
| Cargo recognition | Direct E3 ligase-substrate interaction [10] | Via cargo receptors (p62, NBR1, OPTN, NDP52) [13] [14] |
| Key regulatory components | E1, E2, E3 enzymes, proteasome [10] | ATG proteins, cargo receptors, lysosomal enzymes [11] [12] |
Table 2: Selective Autophagy Types and Cargo Recognition Mechanisms
| Selective Autophagy Type | Cargo | Key Receptors | Ubiquitin Dependence | Disease Associations |
|---|---|---|---|---|
| Aggrephagy | Protein aggregates | p62, ALFY, NBR1 [13] | Yes (K63-linked ubiquitin) [13] | Neurodegenerative diseases (PD, AD, MSA) [15] |
| Pexophagy | Peroxisomes | p62 [13] | Yes | Metabolic disorders [13] |
| Lysophagy | Damaged lysosomes | Not specified | Yes | Lysosomal storage disorders [16] |
| Mitophagy | Damaged mitochondria | BNIP3L/Nix, FUNDC1, OPTN [13] | Varies (Ub-dependent and independent) | Parkinson's disease, metabolic diseases [13] |
| Chaperone-mediated autophagy | Soluble proteins with KFERQ motif | Hsc70, LAMP2A [15] [17] | No | Neurodegenerative diseases, aging [17] |
The Molecular Machinery of Selective Autophagy
Core Autophagy-Related (ATG) Protein Complexes
The initiation and execution of autophagy depend on the coordinated action of core ATG proteins that form functional complexes. The ATG1/ATG13 protein kinase complex acts as the most upstream regulator in response to nutrient status [11]. Under normal conditions, target of rapamycin (TOR) kinase phosphorylates ATG13, reducing its affinity for ATG1, thereby inhibiting autophagy. During stress, TOR inactivation leads to ATG13 dephosphorylation, promoting complex formation with ATG1, ATG11, and ATG101, which initiates phagophore formation [11].
The ATG9/2/18 transmembrane complex provides membrane sources for forming pre-autophagosomal structures, with ATG9 being the only transmembrane protein in the autophagy process [11]. The phosphatidylinositol-3-kinase (PI3K) complex, comprising ATG6, ATG14, VPS15, and VPS34, mediates vesicle nucleation and generates phosphatidylinositol-3-phosphate (PI3P), which recruits additional ATG proteins to the developing phagophore [11].
Two ubiquitin-like conjugation systems are essential for autophagosome formation: the ATG5-ATG12 system and the ATG8-phosphatidylethanolamine (PE) system. ATG12 is activated by ATG7 (E1-like), transferred to ATG10 (E2-like), and conjugated to ATG5. The ATG12-ATG5 complex then interacts with ATG16 to form an E3-like complex that promotes ATG8-PE conjugation [11]. ATG8 (LC3 in mammals) is processed by ATG4 protease before conjugation with PE, a crucial step for autophagosome membrane expansion and closure [11].
Figure 1: Core Autophagy Pathway. Cellular stress inactivates TOR kinase, leading to ULK complex activation and phagophore initiation. The phagophore expands and closes to form an autophagosome, which fuses with lysosomes for cargo degradation.
Cargo Receptors: The Selectivity Specialists
Selective autophagy employs cargo receptors that recognize specific substrates and link them to the core autophagy machinery. p62/SQSTM1 represents the prototypical autophagy receptor, containing multiple domains that enable its function as a scaffolding protein [13]. The PB1 domain facilitates p62 oligomerization, while the ZZ domain interacts with RIP1 in inflammatory signaling. The LC3-interacting region (LIR) mediates direct binding to ATG8/LC3 family proteins, targeting cargo to autophagosomes, and the ubiquitin-associated (UBA) domain recognizes ubiquitinated substrates [13].
Post-translational modifications regulate p62 function. Phosphorylation of serine 403 in the UBA domain by TBK1 and casein kinase 2 enhances its binding affinity for polyubiquitin chains, facilitating efficient cargo recognition [13]. The Keap1-interacting region (KIR) enables p62 to interact with Keap1, leading to NRF2 activation and antioxidant response induction [13].
Other important autophagy receptors include neighbor of BRCA1 gene (NBR1), which shares structural similarities with p62; optineurin (OPTN), involved in mitophagy and aggrephagy; NDP52/CALCOCO2, which targets bacteria and mitochondria; and BNIP3L/Nix and FUNDC1, specialized for mitophagy [13]. These receptors ensure precise cargo selection by recognizing specific degradation signals, often ubiquitin tags, and recruiting the autophagic machinery through interactions with LC3 family proteins.
Figure 2: p62-Mediated Cargo Recognition in Selective Autophagy. Ubiquitinated cargo is recognized by p62 via its UBA domain, while simultaneous interaction with LC3 on the autophagosome membrane through the LIR motif targets cargo for degradation.
Experimental Approaches: Methodologies for Studying ALP
Protocol 1: Monitoring Selective Autophagy via p62 Function
Objective: Assess p62-mediated selective autophagy through aggrephagy analysis [13].
Materials:
- Cell lines (e.g., SH-SY5Y, HeLa)
- p62/SQSTM1 antibodies
- Proteasome inhibitors (MG132, bortezomib)
- Autophagy inhibitors (bafilomycin A1, chloroquine)
- Lysotracker Red for lysosomal staining
- LC3 antibodies
- Ubiquitin antibodies
Methodology:
- Induce protein aggregation by treating cells with proteasome inhibitors (10μM MG132) for 12 hours
- Fix cells and perform immunofluorescence using anti-p62 and anti-ubiquitin antibodies
- Quantify p62-positive aggregates per cell using image analysis software
- Monitor autophagic flux by comparing p62 levels with and without lysosomal inhibitors
- Assess colocalization of p62 with LC3 and ubiquitin using confocal microscopy
- Perform co-immunoprecipitation to analyze p62 interactions with LC3 and ubiquitinated proteins
Expected Results: Under proteasome inhibition, p62 should form puncta that colocalize with ubiquitin and LC3, indicating functional aggrephagy. Lysosomal inhibition should increase these aggregates, demonstrating autophagic flux.
Protocol 2: sEV Retrieval for Autophagy Cargo Analysis
Objective: Isolate and analyze internalized small extracellular vesicles (sEVs) to study selective autophagy mechanisms [18].
Materials:
- MCF7 and MDA-MB-436 cell lines
- Differential ultracentrifuge
- PKH67 fluorescent dye or click chemistry labeling reagents
- Nanoparticle Tracking Analysis system
- Triton X-100 hypotonic solution
- RIPA buffer with protease inhibitors
Methodology:
- Isolate sEVs from conditioned media via differential ultracentrifugation
- Label sEVs with fluorescent markers using PKH67 or click chemistry
- Incubate recipient cells with labeled sEVs for 24-48 hours
- Wash cells thoroughly to remove uninternalized sEVs
- Detach cells and lyse using hypotonic solution
- Perform sequential centrifugation to remove large particles
- Isulate internalized sEVs via ultracentrifugation at 100,000×g for 90 minutes
- Analyze retrieved sEVs using nanoparticle tracking and immunoblotting
Expected Results: Recipient cells selectively internalize specific sEV subpopulations based on functional requirements, demonstrating precision in autophagy-related cargo recognition [18].
Protocol 3: Assessing Ubiquitin-Proteasome and Autophagy Crosstalk
Objective: Evaluate compensatory activation between UPS and ALP using sequential inhibition [10].
Materials:
- Proteasome inhibitors (MG132, bortezomib)
- Autophagy inhibitors (bafilomycin A1, 3-MA)
- Ubiquitin reference proteins
- Antibodies for ubiquitin, p62, LC3, and proteasome subunits
- Cycloheximide to block new protein synthesis
Methodology:
- Treat cells with proteasome inhibitors (5μM MG132) for 8 hours
- Subsequently treat with autophagy inhibitors (100nM bafilomycin A1) for 4 hours
- Harvest cells at different time points for Western blot analysis
- Measure accumulation of polyubiquitinated proteins
- Monitor p62 and LC3-II flux as autophagy indicators
- Assess cell viability and apoptosis markers
- Use cycloheximide to distinguish between newly synthesized and existing proteins
Expected Results: Sequential inhibition should cause synergistic accumulation of ubiquitinated proteins and cellular toxicity, demonstrating functional compensation between degradation systems.
Table 3: Research Reagent Solutions for ALP-UPS Studies
| Reagent/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Inhibitors | MG132, Bortezomib | Proteasome inhibition [10] | UPS blockade to study compensatory autophagy |
| Bafilomycin A1, Chloroquine | Lysosomal inhibition [13] | Autophagic flux measurement | |
| Antibodies | Anti-p62/SQSTM1 | Detect autophagy receptor [13] | Aggrephagy monitoring, protein aggregation studies |
| Anti-LC3 | Autophagosome marker [11] | Autophagic flux assessment, vesicle quantification | |
| Anti-ubiquitin | Detect ubiquitinated substrates [13] | Protein aggregation analysis, UPS-ALP crosstalk | |
| Fluorescent Markers | Lysotracker Red | Lysosomal staining [12] | Lysosomal function and acidity assessment |
| PKH67, Click chemistry | Vesicle and cargo labeling [18] | sEV tracking, uptake studies | |
| Cell Lines | SH-SY5Y | Neurodegeneration models [15] | Protein aggregation, neuroprotection studies |
| MCF7, MDA-MB-436 | Cancer cell models [18] | Selective autophagy, therapeutic response | |
| Expression Systems | GFP-LC3 | Autophagosome visualization [11] | Live-cell imaging of autophagic flux |
| Mutant ubiquitin constructs | Pathway specificity studies [13] | Ubiquitin chain-type function analysis |
Discussion: Therapeutic Implications and Future Directions
The intricate relationship between UPS and ALP presents promising therapeutic opportunities. As these systems functionally complement each other, their coordinated manipulation offers strategies for conditions characterized by proteostasis dysfunction. In neurodegenerative diseases including Alzheimer's, Parkinson's, and multiple system atrophy (MSA), protein aggregation results from impaired clearance rather than excessive production [15] [17]. Both systems show age-related decline, with reduced expression of autophagy-related genes (ATG5, ATG7, BECN1) and decreased proteasomal activity [17].
Emerging therapeutic approaches include autophagy enhancers like rapamycin analogs that inhibit mTOR and activate autophagy [11]. Additionally, targeted protein degradation technologies represent a revolutionary approach. Proteolysis-targeting chimeras (PROTACs) harness UPS for targeted protein degradation, while autophagy-targeting chimeras (AUTACs), autophagosome tethering compounds (ATTEC), and lysosome-targeting chimeras (LYTACs) leverage autophagy for specific protein clearance [12]. These technologies offer particular promise for degrading pathological protein aggregates in neurodegenerative diseases and oncoproteins in cancer [12].
Future research should focus on developing more precise modulators of selective autophagy types, understanding tissue-specific differences in degradation pathways, and identifying biomarkers to monitor pathway activity in human patients. The continued elucidation of molecular mechanisms underlying cargo recognition and degradation will undoubtedly yield novel therapeutic strategies for a wide range of diseases associated with proteostasis dysfunction.
The autophagy-lysosome pathway has evolved from a perceived non-selective bulk degradation system to a highly sophisticated mechanism for specific cargo recognition and clearance. Its complex relationship with the ubiquitin-proteasome system—encompassing both competition and collaboration—creates a robust network for maintaining cellular proteostasis. The discovery of cargo receptors like p62 that recognize ubiquitinated substrates and link them to the autophagic machinery has been particularly transformative, revealing molecular bridges between these degradation pathways.
Continued investigation into the molecular mechanisms of selective autophagy, coupled with advanced technologies for monitoring and manipulating these pathways, holds tremendous promise for understanding and treating human diseases. As we deepen our knowledge of how cells recognize and degrade damaged components, we move closer to developing effective therapies for neurodegenerative diseases, cancer, and other conditions where protein homeostasis is compromised.
Maintaining cellular proteostasis requires precise degradation of misfolded, damaged, or superfluous proteins. This crucial function is primarily executed by two evolutionarily conserved systems: the Ubiquitin-Proteasome System (UPS) and autophagy. While both pathways ultimately target proteins for destruction, they employ fundamentally different molecular machinery and serve complementary cellular roles [1] [19]. The UPS predominantly degrades short-lived regulatory proteins and soluble misfolded proteins through an elaborate enzymatic cascade centered on E1, E2, and E3 enzymes, culminating in proteasomal degradation [19]. In contrast, autophagy specializes in recycling long-lived proteins, damaged organelles, and protein aggregates through the coordinated action of autophagy-related (ATG) proteins and LC3 family proteins, leading to lysosomal degradation [1] [20]. Understanding the distinct components and mechanisms of these systems is paramount for developing targeted therapeutic interventions for neurodegenerative diseases, cancer, and other proteinopathies [19] [21].
Molecular Machinery of the Ubiquitin-Proteasome System (UPS)
The UPS is a sophisticated, ATP-dependent proteolytic system that ensures selective degradation of target proteins with remarkable precision. Its operational framework can be divided into two main phases: ubiquitin conjugation and proteasomal degradation.
The Ubiquitin Conjugation Cascade
Ubiquitination involves a sequential enzymatic cascade that tags substrate proteins with ubiquitin molecules for recognition and degradation by the proteasome:
- E1 (Ubiquitin-Activating Enzyme): This initial enzyme activates ubiquitin in an ATP-dependent reaction, forming a high-energy thioester bond between its catalytic cysteine residue and the C-terminal glycine of ubiquitin. Cells typically express only one or two E1 enzymes, creating a bottleneck in the pathway [20] [19].
- E2 (Ubiquitin-Conjugating Enzyme): Activated ubiquitin is transferred from E1 to the catalytic cysteine of an E2 enzyme, forming an E2~ubiquitin thioester intermediate. The human genome encodes approximately 40 E2 enzymes, which begin to impart substrate diversity [20] [21].
- E3 (Ubiquitin Ligase): This final enzyme in the cascade confers substrate specificity by recognizing target proteins and facilitating the transfer of ubiquitin from E2 to a lysine residue on the substrate. E3 ligases constitute the most diverse UPS component, with humans possessing 600-700 E3s categorized into three major families: RING (Really Interesting New Gene), HECT (Homologous to E6-AP C-Terminus), and RBR (Ring-Between-Ring) ligases [20] [19] [21].
Table 1: Core Enzymatic Components of the Ubiquitin-Proteasome System
| Component | Key Function | Human Genes | Representative Types | Mechanistic Role |
|---|---|---|---|---|
| E1 Enzyme | Ubiquitin activation | 1-2 | UBA1, UBA6 | ATP-dependent ubiquitin C-terminal adenylation and thioester formation |
| E2 Enzyme | Ubiquitin conjugation | ~40 | UBC2, UBC3, UBC4 | Thioester intermediate formation with ubiquitin; determines ubiquitin chain topology |
| E3 Ligase | Substrate recognition & ubiquitin transfer | 600-700 | RING, HECT, RBR, SCF complex | Binds specific substrates and E2~Ub; determines degradation specificity |
| Proteasome | Substrate degradation | ~30 subunits | 19S regulatory particle, 20S core particle | Recognizes polyubiquitinated proteins, unfolds, and proteolytically cleaves them |
Proteasomal Degradation
The 26S proteasome serves as the catalytic endpoint of the UPS, comprising a 20S core particle flanked by two 19S regulatory particles. The 19S particle recognizes polyubiquitinated substrates (typically Lys48-linked chains), deubiquitinates them, unfolds the polypeptide chain in an ATP-dependent manner, and translocates the linearized substrate into the proteolytic chamber of the 20S core particle [1] [19]. Within this chamber, the substrate is cleaved into small peptides by three distinct proteolytic activities: chymotrypsin-like, trypsin-like, and caspase-like activities [19].
Molecular Machinery of Autophagy
Autophagy is a catabolic process that delivers cytoplasmic components to lysosomes for degradation. Macroautophagy (hereafter autophagy) involves the formation of a double-membraned autophagosome that engulfs cargo and fuses with lysosomes. The core autophagy machinery centers on two ubiquitin-like conjugation systems that mediate autophagosome formation.
The ATG12 Conjugation System
The ATG12 system operates analogously to the ubiquitin system but utilizes different conjugation machinery:
- ATG7: Functions as an E1-like enzyme, activating both ATG12 and LC3/ATG8 through thioester bond formation [20] [22].
- ATG10: Serves as the E2-like enzyme specifically for ATG12, transferring activated ATG12 to its target [20].
- ATG5: The acceptor protein for ATG12, forming a stable isopeptide bond [20] [22].
- ATG16L1: Binds noncovalently to the ATG12-ATG5 conjugate, forming a multimeric E3-like complex that facilitates LC3/ATG8 lipidation [23] [20].
The LC3/ATG8 Conjugation System
The second ubiquitin-like system mediates the conjugation of LC3/ATG8 to phosphatidylethanolamine (PE) on the growing phagophore:
- ATG4: A cysteine protease that cleaves the C-terminus of LC3/ATG8 to expose a glycine residue, serving as a priming step [20].
- ATG7: The same E1-like enzyme used in the ATG12 system activates LC3/ATG8 [20] [22].
- ATG3: Functions as the E2-like enzyme specifically for LC3/ATG8 conjugation [20] [22].
- ATG12-ATG5-ATG16L1 Complex: Acts as an E3-like ligase promoting the transfer of LC3/ATG8 from ATG3 to PE, facilitating membrane tethering and hemifusion [23] [20].
Table 2: Core Protein Components of the Autophagy Machinery
| Component | Key Function | System Association | Mechanistic Role | Functional Analogue in UPS |
|---|---|---|---|---|
| ATG7 | E1-like enzyme | Both conjugation systems | Activates ATG12 and LC3/ATG8 via thioester formation | E1 Ubiquitin-Activating Enzyme |
| ATG10 | E2-like enzyme | ATG12 system | Transfers ATG12 to ATG5 | E2 Ubiquitin-Conjugating Enzyme |
| ATG3 | E2-like enzyme | LC3/ATG8 system | Transfers LC3/ATG8 to phosphatidylethanolamine | E2 Ubiquitin-Conjugating Enzyme |
| ATG12-ATG5-ATG16L1 | E3-like complex | Both systems | Promotes LC3/ATG8 lipidation; determines membrane localization | E3 Ubiquitin Ligase |
| LC3/ATG8 | Ubiquitin-like protein | LC3/ATG8 system | Lipid conjugation; autophagosome membrane marker; cargo recruitment | Ubiquitin |
| WIPI2 | Phosphoinositide effector | Phagophore nucleation | Recruits ATG12-ATG5-ATG16L1 to phagophore | N/A |
Comparative Analysis: Key Distinctions and Functional Relationships
While both systems utilize conjugation machinery, their operational principles, substrate preferences, and degradation mechanisms differ significantly.
System-Level Comparison
Table 3: Comparative Analysis of UPS and Autophagy Systems
| Parameter | Ubiquitin-Proteasome System (UPS) | Autophagy |
|---|---|---|
| Primary Substrates | Short-lived regulatory proteins, soluble misfolded proteins [19] | Long-lived proteins, protein aggregates, damaged organelles [1] [19] |
| Degradation Site | 26S Proteasome (cytosol/nucleus) [1] | Lysosome/Vacuole (via autophagolysosome) [1] |
| Energy Requirements | ATP-dependent (ubiquitination & proteasomal degradation) [19] | ATP-dependent (autophagosome formation & fusion) [20] |
| Conjugation Systems | Single ubiquitin system (E1-E2-E3) [19] | Two ubiquitin-like systems (ATG12 & LC3/ATG8) [20] [22] |
| Membrane Involvement | Not required for degradation | Essential (phagophore nucleation, elongation, and fusion) [23] [20] |
| Selectivity Mechanism | E3 ligase-substrate recognition [19] | Receptor-mediated (p62, NBR1, OPTN) via LIR-AIM interactions [1] [20] |
| Ubiquitin Dependence | Essential (K48-linked chains as degradation signal) [1] [19] | Optional (K63-linked chains for selective autophagy) [1] [20] |
| Degradation Products | Small peptides (6-12 amino acids) [19] | Amino acids, fatty acids, nucleotides [19] |
Crosstalk and Coordination
Despite their distinct mechanisms, the UPS and autophagy exhibit significant crosstalk and coordinate to maintain cellular proteostasis:
- Shared Signals: Ubiquitin serves as a common degradation signal, with K48-linked chains typically targeting substrates to the proteasome, while K63-linked chains often mark cargo for autophagic degradation via receptors like p62, NBR1, and OPTN [1].
- Compensatory Activation: Inhibition of one pathway often upregulates the other, demonstrating functional compensation [1].
- Component Degradation: Autophagy can degrade proteasomes (proteaphagy) under certain conditions, while the UPS regulates some autophagy components [1].
- Bridging Molecules: Proteins like EI24 and chaperones (CHIP, BAG1, BAG3) help determine substrate routing between the pathways [1].
Experimental Approaches and Research Reagents
Studying these degradation pathways requires specific methodological approaches and specialized reagents.
Key Experimental Protocols
UPS Activity Assessment:
- Methodology: Treat cells with proteasome inhibitors (MG132, bortezomib) and monitor accumulation of ubiquitinated proteins via western blotting or use fluorescent reporters with degradation signals [24] [19].
- Substrate Tracking: Express model substrates with engineered degrons and measure degradation kinetics using cycloheximide chase assays [24].
- Proteasomal Activity Assays: Use fluorogenic peptides that release fluorescent upon proteasomal cleavage to measure chymotrypsin-like, trypsin-like, and caspase-like activities [19].
Autophagy Flux Measurement:
- LC3 Turnover Assay: Monitor LC3-I to LC3-II conversion by western blot in presence/absence of lysosomal inhibitors (bafilomycin A1, chloroquine) [24] [25].
- Autophagosome Tracking: Express GFP-LC3 and quantify puncta formation microscopically; use tandem mRFP-GFP-LC3 to track autophagosome-lysosome fusion [20] [25].
- Long-Lived Protein Degradation: Measure release of radioactive amino acids from pre-labeled proteins during starvation or stress conditions [19].
Essential Research Reagents
Table 4: Key Research Reagents for Studying UPS and Autophagy
| Reagent | Primary Function | Application | Experimental Readout |
|---|---|---|---|
| MG132 | Proteasome inhibitor | UPS inhibition | Accumulation of polyubiquitinated proteins; reduced degradation of UPS substrates [24] |
| Bafilomycin A1 | V-ATPase inhibitor (blocks lysosomal acidification) | Autophagy flux measurement | Accumulation of LC3-II and autophagosomes; impaired substrate degradation [24] |
| 3-Methyladenine (3-MA) | Class III PI3K inhibitor | Autophagy initiation inhibition | Reduced LC3 lipidation; decreased autophagosome formation [25] |
| Cycloheximide | Protein synthesis inhibitor | Protein half-life measurement | Quantification of substrate degradation kinetics in chase assays [24] |
| Anti-Ubiquitin Antibodies | Detect ubiquitinated proteins | UPS activity assessment | Western blot detection of polyubiquitin chains; immunofluorescence [19] |
| Anti-LC3/ATG8 Antibodies | Detect autophagosomes | Autophagy induction measurement | LC3-I to LC3-II conversion; puncta formation by microscopy [20] [25] |
| GFP-Ubiquitin Reporters | Visualize protein ubiquitination | Real-time UPS monitoring | Live-cell imaging of ubiquitin dynamics; FRET-based sensors [19] |
| Tandem mRFP-GFP-LC3 | Distinguish autophagosomes vs. autolysosomes | Autophagy flux tracking | GFP quenching in acidic lysosomes; red-only signals indicate autolysosomes [20] |
Therapeutic Implications and Research Applications
The distinct but complementary nature of UPS and autophagy pathways presents multiple therapeutic opportunities:
- Targeted Protein Degradation: Bifunctional molecules like PROTACs (Proteolysis-Targeting Chimeras) hijack E3 ubiquitin ligases to degrade disease-causing proteins, while AUTACs (Autophagy-Targeting Chimeras) and ATTECs (Autophagosome-Tethering Compounds) leverage autophagy for targeted degradation [26] [21].
- Neurodegenerative Diseases: Enhancing autophagy flux shows promise for clearing aggregation-prone proteins in Alzheimer's (Aβ, tau), Parkinson's (α-synuclein), and Huntington's disease (mutant huntingtin) [19].
- Cancer Therapeutics: Proteasome inhibitors (bortezomib, carfilzomib) are FDA-approved for multiple myeloma, while autophagy modulation (inhibition or enhancement) shows potential in various cancer contexts [19].
- Aging and Senescence: Both systems decline with age; NMD impairment via reduced UPF1 levels contributes to cellular senescence, highlighting the interconnectedness of degradation pathways in aging [24].
The ubiquitin-proteasome system and autophagy represent two fundamental pillars of cellular protein degradation with distinct yet interconnected molecular components. The E1/E2/E3 enzyme cascade of the UPS provides rapid, selective degradation of individual proteins, while the ATG/LC3 protein machinery of autophagy enables bulk clearance of larger structures and aggregates. Their sophisticated coordination maintains proteostatic balance, and dysregulation in either system contributes to numerous diseases. Continued comparative analysis of these pathways will undoubtedly yield novel therapeutic strategies for cancer, neurodegenerative disorders, and other proteinopathies.
Eukaryotic cells employ two primary systems for protein degradation: the Ubiquitin-Proteasome System (UPS) and autophagy. While both are essential for cellular homeostasis, they utilize distinct mechanisms for substrate recognition. The UPS is characterized by its rapid degradation of short-lived and soluble proteins, with K48-linked polyubiquitin chains serving as the principal signal for proteasomal destruction [27] [28]. In contrast, selective autophagy eliminates larger structures such as protein aggregates and damaged organelles, relying on LC3-interacting region (LIR) motifs, also known as Atg8-interacting motifs (AIMs), to tether cargo to the growing autophagosome [29] [28]. This guide provides a detailed comparison of these recognition systems, supported by experimental data and methodologies relevant to research on misfolded protein degradation.
The following table summarizes the core characteristics of the two signal recognition systems.
Table 1: Comparative Analysis of K48-Ubiquitin and LIR/AIM Motif Systems
| Feature | K48-Ubiquitin in UPS | LIR/AIM in Selective Autophagy |
|---|---|---|
| Primary Function | Recognition signal for proteasomal degradation [27] [28] | Cargo tethering to the phagophore via binding to Atg8-family proteins (LC3/GABARAP) [29] [30] |
| System Role | Degradation of short-lived, soluble proteins (e.g., cell cycle regulators, misfolded proteins) [27] | Selective degradation of bulky cargo (e.g., protein aggregates, organelles, pathogens) [28] [31] |
| Key Players | E1/E2/E3 enzymes, Proteasome (19S cap subunits Rpn10/Rpn13), K48-ubiquitin chains [27] | Cargo receptors (e.g., p62, NBR1, OPTN), Atg8-family proteins, LIR/AIM motif [29] [28] |
| Signal Nature | Covalent post-translational modification [27] | Modular protein-protein interaction motif [29] |
| Structural Basis | Polyubiquitin chain linked via Lys48 residue; recognized by UBDs in proteasomal subunits [27] [28] | Conserved core sequence [W/F/Y]-X-X-[L/I/V]; binds hydrophobic pockets in LIR docking site (LDS) of Atg8 proteins [29] |
K48-Ubiquitin Recognition by the Ubiquitin-Proteasome System
Pathway Mechanism
The UPS is the main pathway for targeted protein turnover. The K48-linked polyubiquitin chain is the canonical signal that directs substrates to the 26S proteasome for degradation [27] [28]. This process involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that attach a chain of ubiquitin molecules linked through lysine 48 (K48) to a target protein [27]. The 26S proteasome recognizes this chain via ubiquitin receptors (e.g., Rpn10 and Rpn13) in its 19S regulatory cap. The substrate is then unfolded, deubiquitinated, and translocated into the 20S core proteasome for proteolytic digestion [27].
Diagram: K48-Ubiquitin Proteasomal Degradation Pathway
Key Experimental Protocols
1. In Vitro Ubiquitination Assay: This protocol reconstitutes the ubiquitination cascade to study E3 ligase specificity and chain linkage formation [27].
- Methodology: Incubate the purified target protein with E1 enzyme, specific E2 enzyme, E3 ligase, ubiquitin, and an ATP-regenerating system in a suitable reaction buffer.
- Analysis: Terminate the reaction at time points with SDS sample buffer. Analyze ubiquitination by western blotting using an antibody against the target protein to observe higher molecular weight smears, or against ubiquitin. To confirm K48-linkage, use ubiquitin mutants (K48R, which cannot form K48 chains) or linkage-specific antibodies (e.g., anti-K48-ubiquitin) [28] [32].
2. Proteasomal Degradation Assay: This measures the fate of the ubiquitinated protein.
- Methodology: Incubate a radioactively or fluorescently labeled substrate protein (ubiquitinated in vitro or in cells) with purified 26S proteasomes in degradation buffer (containing ATP). To confirm proteasome-dependence, include a control with the proteasome inhibitor MG132.
- Analysis: Monitor the disappearance of the full-length substrate over time by techniques like SDS-PAGE and autoradiography/fluorography, or by measuring the release of trichloroacetic acid-soluble counts [27].
LIR/AIM Motif Recognition in Selective Autophagy
Pathway Mechanism
Selective autophagy relies on a family of cargo receptors (e.g., p62, NBR1, OPTN) that physically bridge the cargo to the autophagosomal membrane. This bridge is formed when a conserved LIR/AIM motif in the receptor binds to Atg8-family proteins (e.g., LC3, GABARAP) that are lipidated to the phagophore membrane [29] [28]. The canonical LIR motif is a short, degenerate sequence with the core pattern [W/F/Y]-X-X-[L/I/V], where X is any amino acid [29]. This motif docks into a hydrophobic pocket called the LIR docking site (LDS) on the surface of Atg8-family proteins, thereby enclosing the cargo within the forming autophagosome [29].
Diagram: LIR/AIM-Mediated Selective Autophagy Pathway
Key Experimental Protocols
1. LIR Motif Identification and Validation: This protocol identifies and confirms functional LIR motifs within a candidate protein [29].
- Bioinformatic Screening: Scan the protein's amino acid sequence for patterns matching the canonical LIR consensus ([W/F/Y]-X-X-[L/I/V]).
- Pull-Down/Co-Immunoprecipitation (Co-IP): Incubate a purified protein fragment containing the putative LIR motif with purified Atg8-family proteins (e.g., LC3). Alternatively, express the full-length candidate protein in cells and perform Co-IP with antibodies against endogenous LC3. A critical control is to introduce point mutations (e.g., substituting the conserved aromatic residue with Ala) in the putative LIR motif, which should abolish binding [29].
- Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC): Use these biophysical methods with synthesized LIR peptides and purified Atg8 proteins to quantitatively measure the binding affinity (KD) [29].
2. Functional Autophagy Cargo Assay: This assesses whether the LIR-dependent binding drives actual cargo degradation via autophagy.
- Methodology: Express a fluorescently tagged cargo protein (e.g., a mutant protein that forms aggregates) along with wild-type or LIR-mutated receptor in cells. Induce autophagy (e.g., by starvation or mTOR inhibition) and track cargo localization and clearance.
- Analysis: Use immunofluorescence microscopy to monitor the co-localization of the cargo, the receptor, and LC3 puncta. Quantify cargo clearance by western blotting or flow cytometry. Inhibition of autophagy with drugs like Bafilomycin A1 (which blocks lysosomal degradation) should lead to cargo accumulation, confirming an autophagic route [33] [28].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Studying K48-Ubiquitin and LIR/AIM Pathways
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| K48-Specific Ubiquitin Antibody | Detects and validates K48-linked polyubiquitin chains [28] [34] | Immunoblotting, immunofluorescence after proteasomal inhibition. |
| Ubiquitin Mutants (K48R, K63R) | Determines chain linkage specificity in vitro and in vivo [34] [32] | Expressing K48R Ub mutant prevents K48-chain formation, used to validate signal. |
| Proteasome Inhibitors (MG132, Bortezomib) | Blocks proteasomal activity to study UPS substrate accumulation [27] | Validates if protein degradation is proteasome-dependent. |
| Recombinant Atg8-Family Proteins (LC3B, GABARAP) | In vitro binding studies to map and quantify LIR/AIM interactions [29] [30] | Pull-down assays, SPR, ITC with candidate LIR peptides. |
| Tandem Fluorescent-LC3 (e.g., mRFP-GFP-LC3) | Monitors autophagic flux in live cells [34] | GFP signal quenched in acidic lysosomes, while mRFP is stable; red-only puncta indicate degradative flux. |
| LIR-Mutant Constructs | Negative control to confirm LIR-dependent processes [29] | Mutating core LIR residues (e.g., W to A) disrupts receptor-Atg8 binding and blocks selective autophagy. |
Integrated Crosstalk and Research Implications
While distinct, the UPS and autophagy pathways are interconnected. Impairment of the UPS can often induce autophagy as a compensatory degradation mechanism [27] [32]. Furthermore, ubiquitin itself serves as a common signal: many substrates of selective autophagy are first ubiquitinated, and autophagy receptors like p62 and NBR1 contain both a LIR motif to bind LC3 and a ubiquitin-associated (UBA) domain to bind ubiquitin on the cargo, thereby linking the two systems [28] [35]. A key example is the E3 ligase Smurf1, which facilitates the autophagy of Mycobacterium tuberculosis by decorating the pathogen with ubiquitin chains, enabling recognition by autophagy receptors [33].
Understanding these specific recognition signals is vital for drug development. Targeting specific E3 ligases or the LIR-Agt8 interface offers potential for therapeutic intervention in diseases characterized by proteostasis failure, such as neurodegenerative disorders and cancer. The experimental frameworks and tools outlined here provide a foundation for advancing research in this critical field.
Physiological Roles of UPS and Autophagy in Cellular Proteostasis
Cellular proteostasis, the delicate balance of protein synthesis, folding, and degradation, is fundamental to cellular health and function. Two primary degradation systems maintain this balance: the ubiquitin-proteasome system (UPS) and autophagy. The ubiquitin-proteasome system is a selective, rapid-response mechanism that identifies and degrades short-lived proteins and soluble misfolded proteins, acting as a precision tool for cellular cleanup [36]. In contrast, autophagy (specifically macroautophagy) is a bulk degradation process that eliminates long-lived proteins, insoluble protein aggregates, and damaged organelles through lysosomal degradation, serving as the cell's bulk recycling plant [36] [37]. While traditionally viewed as independent pathways, emerging research reveals sophisticated coordination between these systems in managing proteotoxic stress, with implications for understanding disease mechanisms and developing novel therapeutic strategies [38] [39].
The critical importance of these proteostasis networks becomes evident during cellular stress. Accumulation of misfolded proteins in the endoplasmic reticulum (ER) triggers the unfolded protein response (UPR), which dynamically orchestrates adaptive responses by modulating both UPS and autophagy functions [40] [37]. When ER stress becomes irremediable, the UPR shifts signaling toward apoptosis, demonstrating the life-or-death decisions mediated by these interconnected quality control systems [40]. This comparison guide examines the physiological roles, molecular mechanisms, and experimental approaches for studying UPS and autophagy, providing researchers with a structured framework for understanding their complementary functions in cellular proteostasis.
The UPS and autophagy operate through distinct yet interconnected mechanisms to maintain proteostasis. The UPS functions as the primary pathway for targeted protein degradation, relying on a cascade of enzymatic reactions: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligating) enzymes that tag substrate proteins with polyubiquitin chains, predominantly through K48 linkages, marking them for destruction by the 26S proteasome [36]. This system excels at processing short-lived regulatory proteins and soluble misfolded proteins with remarkable specificity and speed.
Autophagy encompasses several subtypes, with macroautophagy being the primary pathway for bulk degradation of cytoplasmic components. This process involves the formation of double-membrane vesicles called autophagosomes that engulf cargo, which then fuse with lysosomes for content degradation [36] [37]. Selective forms of autophagy target specific cargoes: mitophagy for damaged mitochondria, reticulophagy for ER fragments, and aggrephagy for protein aggregates [38] [37]. The autophagy pathway is particularly crucial for eliminating organelles and protein aggregates that are too large for proteasomal degradation.
Table 1: Fundamental Characteristics of UPS and Autophagy
| Characteristic | Ubiquitin-Proteasome System (UPS) | Autophagy |
|---|---|---|
| Primary Degradation Machinery | 26S proteasome | Lysosome/Autolysosome |
| Main Substrate Types | Short-lived proteins, soluble misfolded proteins [36] | Long-lived proteins, protein aggregates, damaged organelles [36] |
| Ubiquitin Dependence | Essential (K48-linked chains) [36] | Selective (involves ubiquitin-like proteins and receptors) [39] |
| Temporal Profile | Rapid (minutes to hours) | Slower (hours) |
| Specificity | High (enzyme-substrate specific) | Bulk degradation with emerging selective mechanisms |
| Energy Requirements | ATP-dependent | ATP-dependent |
| Key Adaptor Molecules | E3 ubiquitin ligases [36] | p62/SQSTM1, LC3, ATG proteins [39] |
The functional relationship between these systems extends beyond mere redundancy. Research indicates sophisticated crosstalk and compensation mechanisms between UPS and autophagy. When the UPS is compromised, autophagy can be upregulated to alleviate proteotoxic stress, and conversely, inhibition of autophagy can enhance UPS activity [38] [41]. The hub protein p62/SQSTM1 exemplifies this interconnection, functioning as a selective autophagy receptor that recognizes ubiquitinated proteins and targets them for autophagic degradation, thereby serving as a molecular bridge between the two systems [39].
Molecular Mechanisms and Signaling Pathways
The Ubiquitin-Proteasome System Cascade
The UPS operates through a finely tuned enzymatic cascade that begins with ubiquitin activation. An E1 activating enzyme utilizes ATP to form a high-energy thioester bond with ubiquitin, which is then transferred to an E2 conjugating enzyme. The final specificity comes from E3 ubiquitin ligases, which recognize specific substrate proteins and facilitate the transfer of ubiquitin from E2 to the target protein [36]. Repeated cycles result in polyubiquitination, with K48-linked chains serving as the primary signal for proteasomal recognition and degradation [36].
The 26S proteasome itself consists of a 20S core particle that houses the proteolytic active sites and 19S regulatory particles that recognize ubiquitinated proteins, remove ubiquitin chains, unfold the target protein, and translocate it into the core for degradation [36]. This sophisticated machinery ensures precise temporal control of protein abundance, regulating critical cellular processes including cell cycle progression, signal transduction, and gene expression.
Autophagy Pathway and Regulation
Autophagy initiation involves the formation of a phagophore that expands to become an autophagosome. This process is regulated by a conserved set of autophagy-related (ATG) proteins and the autophagy hub protein p62/SQSTM1 [39]. p62 possesses multiple domains that facilitate its function as a selective autophagy receptor: a PB1 domain for self-oligomerization, a UBA domain for binding ubiquitinated proteins, and an LIR motif for interacting with LC3 on developing autophagosomes [39].
The UPR represents a key regulatory node connecting ER stress to autophagy activation. The three UPR sensors - PERK, IRE1α, and ATF6 - detect protein misfolding in the ER and initiate signaling cascades that modulate autophagy through different mechanisms [40] [37]. PERK-mediated phosphorylation of eIF2α attenuates global protein synthesis while selectively promoting ATF4 translation, which activates transcription of autophagy-related genes including p62, Atg7, and Atg5 [37]. IRE1α activation leads to XBP1 splicing and can promote autophagy through JNK signaling, while ATF6 activation increases expression of ER quality control components that direct misfolded proteins toward degradation [40].
Figure 1: UPR Pathway Regulation of Autophagy. ER stress activates three UPR sensors (PERK, IRE1α, ATF6) that initiate signaling cascades modulating autophagy through transcriptional upregulation of autophagy-related genes and ERAD components.
Integrated Proteostasis Network
The coordination between UPS and autophagy extends beyond compensatory activation to include shared regulatory mechanisms. The N-degron pathway has recently been identified as a mediator between these systems, governing the stability of autophagy components like ATG8a through Arg/N-degron recognition by N-recognins such as UBR7, which targets them for proteasomal degradation [42]. This demonstrates how the UPS directly regulates autophagy machinery.
During ischemic/reperfusion injury, p62 emerges as a master regulator that orchestrates both degradation systems, modulating cell fate decisions by balancing autophagy activation with regulation of antioxidant signaling through NRF2 and control of inflammatory responses via NFκB [39]. This integrated network allows cells to tailor degradation capacity to specific proteotoxic challenges, allocating resources between the precision targeting of UPS and the bulk clearance capacity of autophagy.
Experimental Approaches and Methodologies
Quantitative Turnover Mapping
Systematic analysis of protein degradation pathways has been revolutionized by approaches like the Turnover Map (T-MAP), which combines quantitative proteomics with genetic perturbations to deconvolve degradation pathways [41]. This method involves pulse-labeling of yeast or mammalian cells with stable isotope-labeled amino acids, followed by mass spectrometry-based measurement of protein turnover rates across a panel of knockout strains deficient in various components of the degradation machinery.
The T-MAP approach enables researchers to identify whether specific proteins are primarily degraded by proteasomal or vacuolar/lysosomal pathways by analyzing stabilization patterns in mutants affecting each system (e.g., rpn4Δ affecting proteasome function versus pep4Δ affecting vacuolar proteases) [41]. This systematic profiling has revealed that approximately 15% of the yeast proteome consists of short- and medium-lived proteins actively degraded at steady state, enriched for regulatory proteins like transcription factors, kinases, and membrane transporters [41].
Table 2: Experimental Approaches for Studying UPS and Autophagy
| Method Category | Specific Methods | Key Applications | Interpretation Notes |
|---|---|---|---|
| Degradation Inhibition | MG132 (proteasome inhibitor) [42], Concanamycin A (vacuolar ATPase inhibitor) [42], Bafilomycin A1 (autophagy inhibitor) | Pathway assignment, substrate identification | Confirm specificity with multiple inhibitors; monitor compensatory activation |
| Turnover Measurements | Cycloheximide chase [41], SILAC/pulse-chase proteomics [41], T-MAP [41] | Degradation kinetics, pathway mapping | Consider cell type variations; account for protein synthesis effects |
| Genetic Perturbations | CRISPR/Cas9 KO, RNAi, Dominant-negative constructs, Inducible systems [39] | Pathway necessity, functional validation | Monitor adaptive responses; consider compensation between pathways |
| Imaging & Localization | Fluorescence microscopy, Immuno-gold EM, Colocalization studies | Spatial organization, pathway activity | Quantitative analysis required for robust conclusions |
| Activity Reporters | LC3-II lipidation [39], Ubiquitin chain linkage analysis [36], Keima assay | Pathway activity, substrate flux | Use multiple complementary reporters for verification |
Pathway-Specific Functional Assays
For targeted analysis of UPS function, researchers commonly employ substrate-based reporters, ubiquitination assays, and proteasome activity measurements. Immunoblotting for polyubiquitinated proteins and specific ubiquitin linkages (K48 versus K63) can indicate pathway engagement [36]. Monitoring stabilization of known UPS substrates (e.g., ATG8a(L) [42]) in response to proteasome inhibitors like MG132 provides functional validation of UPS dependence.
Autophagy assessment typically involves multiple complementary approaches due to the dynamic nature of the process. LC3 lipidation (LC3-I to LC3-II conversion) and p62 degradation assays monitor autophagic flux when measured with and without lysosomal inhibitors [39]. Imaging-based approaches using GFP-LC3 reporters track autophagosome formation and turnover, while selective autophagy can be assessed by monitoring cargo receptor recruitment and colocalization with autophagosomal markers [39].
Figure 2: Experimental Workflow for Degradation Pathway Analysis. Comprehensive assessment of protein degradation pathways requires a multi-step approach combining pharmacological inhibition, substrate stabilization analysis, genetic validation, and turnover kinetics measurement, with pathway-specific assays for UPS and autophagy.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for UPS and Autophagy Studies
| Reagent/Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Proteasome Inhibitors | MG132 [42], Bortezomib, Carfilzomib | Block proteasome activity, stabilize UPS substrates | Can induce compensatory autophagy; use dose titration |
| Lysosomal Inhibitors | Concanamycin A [42], Bafilomycin A1, Chloroquine | Inhibit lysosomal degradation, measure autophagic flux | Can affect endosomal trafficking; monitor lysosomal pH |
| UPS Activity Reporters | Ubiquitin chain linkage antibodies, Proteasome activity probes | Monitor ubiquitination patterns, proteasome function | Distinguish K48 vs K63 linkages for pathway assignment |
| Autophagy Modulators | Rapamycin, Torin1 (inducers), 3-MA (inhibitor) | Modulate autophagy flux, test pathway necessity | Consider temporal effects; acute vs chronic modulation |
| Critical Antibodies | Anti-p62 [39], Anti-LC3 [39], Anti-Ubiquitin, Anti-K48/K63 linkage | Detect key pathway components, post-translational modifications | Validate species specificity; check phosphorylation status |
| Genetic Tools | shRNA-p62 [39], HA-p62 overexpression [39], CRISPR E3 KO | Pathway manipulation, substrate validation | Use inducible systems for essential genes; monitor adaptation |
| Pathway Reporters | GFP-LC3, Keima assays, Ubiquitin degradation reporters | Visualize pathway activity, monitor substrate degradation | Use tandem fluorescent tags for flux measurements |
Discussion and Research Implications
The sophisticated interplay between UPS and autophagy represents a fundamental aspect of cellular proteostasis with far-reaching implications for understanding disease mechanisms and developing therapeutic interventions. The emerging paradigm reveals not simply redundant systems, but an integrated proteostasis network with distributed responsibilities, compensatory capacities, and shared regulatory nodes [38] [39]. This network exhibits remarkable plasticity, adapting to diverse proteotoxic challenges through dynamic reallocation of degradation resources.
From a therapeutic perspective, the interconnected nature of these pathways presents both challenges and opportunities. Targeted protein degradation technologies, including PROTACs and molecular glues that harness the UPS, and LYTACs and AUTACs that engage lysosomal degradation, represent promising therapeutic avenues [36] [43]. However, the compensatory activation between pathways necessitates careful consideration of potential resistance mechanisms. For example, prolonged proteasome inhibition can upregulate autophagy as an escape mechanism, suggesting that dual-pathway targeting may be required for complete efficacy in certain contexts [38].
Future research directions should focus on quantifying the flux distribution between these pathways under various physiological and pathological conditions, developing more precise tools for real-time monitoring of degradation kinetics in live cells, and identifying master regulators that coordinate pathway allocation. The systematic T-MAP approach [41] provides a framework for such investigations, enabling comprehensive degradation pathway mapping across diverse cell types and disease states. As our understanding of the physiological roles of UPS and autophagy continues to evolve, so too will our ability to therapeutically modulate these essential proteostasis guardians in human disease.
Harnessing Degradation Pathways: From PROTACs to AUTACs in Drug Discovery
Targeted Protein Degradation (TPD) represents a groundbreaking paradigm shift in modern drug discovery and therapeutic development, moving beyond the limitations of traditional occupancy-based inhibitors to an event-driven model that directly eliminates disease-causing proteins [44] [45]. This innovative approach strategically exploits the cell's natural protein quality control machinery—primarily the ubiquitin-proteasome system (UPS)—to achieve precise degradation of specific pathological proteins [36] [46]. Within this rapidly evolving field, two complementary technologies have emerged as frontrunners: PROteolysis TArgeting Chimeras (PROTACs) and Molecular Glue Degraders (MGDs) [44] [47]. Both modalities facilitate the ubiquitination and subsequent proteasomal degradation of proteins of interest (POIs), but they differ fundamentally in their molecular architecture and mechanism of action [45].
The context of degrading misfolded proteins is particularly relevant for neurodegenerative diseases, where the accumulation of toxic protein aggregates is a common pathological feature [44]. While the UPS primarily handles soluble, short-lived proteins, autophagy is responsible for clearing larger structures like protein aggregates and damaged organelles [36]. This guide focuses on UPS-dependent strategies, specifically PROTACs and molecular glues, which offer unprecedented opportunities for targeting "undruggable" proteins that have eluded conventional therapeutic approaches [44] [45].
The Ubiquitin-Proteasome System: Core Machinery for Targeted Degradation
The ubiquitin-proteasome system is a highly regulated, ATP-dependent pathway that maintains cellular protein homeostasis by selectively degrading damaged, misfolded, or short-lived regulatory proteins [36] [46]. This process involves a sequential enzymatic cascade:
- Activation: The E1 activating enzyme activates ubiquitin in an ATP-dependent reaction [46].
- Conjugation: The activated ubiquitin is transferred to an E2 conjugating enzyme [36].
- Ligation: An E3 ubiquitin ligase recognizes specific substrate proteins and facilitates the transfer of ubiquitin from E2 to the target protein [36] [46].
- Degradation: Polyubiquitinated proteins are recognized and degraded by the 26S proteasome into small peptides [46].
E3 ubiquitin ligases are particularly important as they provide substrate specificity, with over 600 known human E3 ligases offering diverse targeting opportunities [36] [48]. The proteasome itself is a multi-subunit complex comprising a 20S core particle responsible for proteolysis, capped by 19S regulatory particles that recognize ubiquitinated substrates and initiate the degradation process [46].
The following diagram illustrates the sequential enzymatic cascade of the ubiquitin-proteasome system, which is hijacked by both PROTACs and molecular glues to achieve targeted protein degradation.
PROTACs: Heterobifunctional Inducers of Protein Degradation
Mechanism of Action and Molecular Architecture
PROTACs are heterobifunctional molecules consisting of three key components: a ligand that binds to the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a chemical linker connecting these two moieties [44] [36]. The mechanism is catalytic—a single PROTAC molecule can facilitate the degradation of multiple POI molecules, as it is not consumed in the degradation process [44] [48]. This catalytic nature allows for potent effects at low concentrations and sustained protein knockdown [44].
The degradation process follows a specific sequence:
- The PROTAC molecule simultaneously binds to both the POI and an E3 ubiquitin ligase [44].
- This forced proximity induces formation of a ternary complex (E3 ligase-PROTAC-POI) [49].
- The E3 ligase transfers ubiquitin chains to lysine residues on the POI [46].
- The polyubiquitinated POI is recognized by the 26S proteasome [46].
- The POI is degraded into small peptide fragments, while the PROTAC is released to catalyze another round of degradation [48].
Quantitative Profiling of PROTAC Characteristics
Table 1: Comparative Analysis of PROTAC Properties and Applications
| Characteristic | PROTAC Profile | Supporting Evidence |
|---|---|---|
| Molecular Weight | High (typically 700-1200 Da) [44] | Creates solubility and permeability challenges [44] |
| Oral Bioavailability | Often challenging [44] | Limited by size and lipophilicity [44] [45] |
| BBB Penetration | More challenging for CNS targets [44] | Large size hinders brain access [44] |
| Discovery Strategy | Rational design framework [44] | Systematic linker optimization between known ligands [44] |
| Catalytic Nature | Event-driven, sub-stoichiometric activity [44] [48] | Single molecule degrades multiple POIs [44] |
| Therapeutic Areas | Oncology, neurodegeneration, autoimmune diseases [44] | ARV-471 (breast cancer) in Phase III trials [44] [50] |
| Key Challenges | Hook effect, poor membrane permeability, off-target effects [44] [48] | High concentrations saturate binding sites [44] |
Molecular Glues: Monovalent Inducers of Protein-Protein Interactions
Mechanism of Action and Molecular Architecture
Molecular glues are monovalent small molecules that induce or stabilize novel protein-protein interactions (PPIs) between an E3 ubiquitin ligase and a target protein, leading to ubiquitination and degradation [44] [47]. Unlike PROTACs, molecular glues are typically single, relatively small molecules (<500 Da) that do not contain a linker [44] [47]. Their mechanism generally involves binding to one protein (often the E3 ligase), inducing a conformational change or creating a "neosurface" that becomes complementary to a specific region on the POI [44]. This effectively "glues" the E3 ligase and POI together into a stable ternary complex, enabling ubiquitination and subsequent degradation [44].
A notable recent example is MRT-31619, a molecular glue that drives homo-dimerization of the CRBN E3 ligase, promoting its fast, potent, and selective degradation [51]. Structural studies revealed that two MRT-31619 molecules assemble into a helix-like structure that drives ternary complex formation by mimicking a neosubstrate G-loop degron [51].
Quantitative Profiling of Molecular Glue Characteristics
Table 2: Comparative Analysis of Molecular Glue Properties and Applications
| Characteristic | Molecular Glue Profile | Supporting Evidence |
|---|---|---|
| Molecular Weight | Lower (typically <500 Da) [44] | Improved drug-like properties [44] |
| Oral Bioavailability | Generally improved [44] | Smaller size enhances absorption [44] |
| BBB Penetration | Generally better for CNS targets [44] | Smaller size facilitates brain access [44] |
| Discovery Strategy | Historically serendipitous; increasingly rational/AI-driven [44] | IMiDs discovered before mechanism understood [44] [36] |
| Catalytic Nature | Event-driven, sub-stoichiometric activity [44] | Similar catalytic mechanism to PROTACs [44] |
| Therapeutic Areas | Oncology, autoimmune disorders, neurodegenerative diseases [44] | IMiDs (multiple myeloma) [44] [36] |
| Key Challenges | Discovery difficulty, limited E3 ligase repertoire [44] | Most target CRBN; rational design challenging [44] |
Direct Comparative Analysis: PROTACs vs. Molecular Glues
Table 3: Side-by-Side Comparison of Key Features Between PROTACs and Molecular Glues
| Feature | PROTACs | Molecular Glues |
|---|---|---|
| Molecular Structure | Bifunctional (heterobifunctional) [44] | Monovalent (single molecule) [44] |
| Linker | Required for connecting two ligands [44] | Linker-less [44] |
| Molecular Weight | Higher (typically 700-1200 Da) [44] | Lower (typically <500 Da) [44] |
| Oral Bioavailability | Often challenging due to size/lipophilicity [44] | Generally improved due to smaller size [44] |
| BBB Penetration | More challenging for CNS targets [44] | Generally better for CNS targets [44] |
| Discovery Strategy | More rational design framework, linker optimization [44] | Historically serendipitous; increasingly rational/AI-driven [44] |
| Mechanism of Action | Brings two pre-existing binding sites into proximity [44] | Induces or stabilizes a new protein-protein interface [44] |
| E3 Ligase Utilization | Can utilize various E3 ligases (CRBN, VHL, MDM2) [44] [36] | Primarily CRBN-focused currently [44] |
| Hook Effect | Observable at high concentrations [44] [48] | Not typically observed (e.g., MRT-31619) [51] |
Experimental Approaches and Research Methodologies
Key Experimental Protocols for Degradation Studies
Protocol 1: Assessing Degradation Efficiency and Kinetics
Objective: Quantify target protein degradation over time to determine degradation efficiency (DC₅₀) and maximum degradation (Dmax) [44].
Methodology:
- Treat cells with varying concentrations of PROTAC or molecular glue for predetermined timepoints (e.g., 4, 8, 24 hours) [44].
- Lyse cells and quantify target protein levels using Western blotting or mass spectrometry-based proteomics [44].
- Measure global proteome changes to assess selectivity and off-target effects [44].
- Calculate DC₅₀ values (concentration achieving 50% degradation) and Dmax (maximum degradation achieved) from dose-response curves [49].
Protocol 2: Ternary Complex Formation Analysis
Objective: Confirm and characterize the formation of E3 ligase-degrader-POI ternary complexes [49].
Methodology:
- Use techniques such as NanoBRET to monitor protein-protein interactions in live cells [51].
- Employ surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) to measure binding affinities and cooperative effects [47].
- Utilize X-ray crystallography or cryo-EM to determine ternary complex structures at atomic resolution [51].
- Perform molecular dynamics simulations to study complex stability and lysine residue positioning for ubiquitination [49].
Protocol 3: Mechanistic Validation through Rescue Experiments
Objective: Confirm that observed degradation occurs via the intended UPS pathway [51].
Methodology:
- Pre-treat cells with proteasome inhibitors (e.g., bortezomib) or NEDD8-activating enzyme inhibitors (e.g., MLN4924) [51].
- Administer the degrader compound and assess whether degradation is blocked [51].
- Use CRISPR/Cas9 to knockout the relevant E3 ligase and test degradation dependence [51].
- Compete with known E3 ligase binders (e.g., lenalidomide for CRBN) to confirm binding site specificity [51].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for TPD Investigations
| Reagent/Solution | Function/Application | Specific Examples |
|---|---|---|
| E3 Ligase Ligands | Recruit specific E3 ligases to degradation complexes [44] | Thalidomide derivatives (CRBN) [44]; VHL ligands [36]; MDM2 ligands (e.g., nutlin-3) [36] |
| Proteasome Inhibitors | Validate UPS-dependent degradation mechanism [51] [46] | Bortezomib, carfilzomib, ixazomib, MG132 [51] [46] |
| NEDD8 Activator Inhibitor | Blocks cullin-RING ligase activation [51] | MLN4924 (inhibits NEDD8 E1 enzyme) [51] |
| Humanized CRBN Models | Evaluate CRBN-recruiting degraders in physiologically relevant systems [50] | B-hCRBN mice (mouse Crbn replaced with human CRBN) [50] |
| Mass Spectrometry Proteomics | Assess degradation selectivity and off-target effects [44] | Next-generation DIA technology for deep protein profiling [44] |
| Structural Biology Tools | Characterize ternary complex formation at atomic level [51] | Cryo-EM, X-ray crystallography [51] |
Visualization of PROTAC and Molecular Glue Mechanisms
The following diagrams illustrate the distinct mechanisms of action for PROTACs versus molecular glues, highlighting how each technology facilitates the ubiquitination and degradation of target proteins.
PROTACs and molecular glues represent complementary approaches within the TPD landscape, each with distinct advantages and limitations. PROTACs offer a more rational design framework and the ability to target numerous proteins with various E3 ligases, but face challenges related to their larger size and physicochemical properties [44] [48]. Molecular glues benefit from superior drug-like properties but have historically been difficult to discover through rational design, though this is changing with advanced screening and computational methods [44] [45].
The field is rapidly evolving with several innovative strategies emerging, including activatable PROTACs triggered in specific cellular environments, dual-targeting PROTACs that degrade multiple proteins simultaneously, and nano-PROTACs that improve delivery and targeting [48]. Additionally, researchers are exploring beyond the traditional UPS by developing lysosome-targeting degraders that could handle different classes of targets, including extracellular proteins and protein aggregates [36] [52].
As the first PROTAC and molecular glue candidates advance through clinical trials, with vepdegestrant (ARV-471) currently in Phase III trials for breast cancer [50], these technologies are poised to fundamentally transform therapeutic development across oncology, neurodegenerative diseases, and inflammatory conditions. Their ability to target previously "undruggable" proteins represents one of the most promising frontiers in modern pharmacology, offering new hope for treating diseases with high unmet medical needs.
The maintenance of cellular proteostasis is fundamental to cell survival and function. For decades, the ubiquitin-proteasome system (UPS) has been recognized as the primary pathway for degrading short-lived intracellular proteins, while the autophagy-lysosomal pathway has been understood to handle bulkier cargo, including protein aggregates and damaged organelles [36] [53]. This traditional division of labor is being redefined by emerging targeted protein degradation (TPD) technologies that strategically exploit these pathways for therapeutic purposes.
Proteolysis-targeting chimeras (PROTACs), which harness the UPS, have demonstrated remarkable success in degrading numerous intracellular proteins and several candidates have entered clinical trials [36]. However, intrinsic limitations of the proteasome, including its narrow catalytic chamber (approximately 13 Å in diameter) and inability to degrade non-proteinaceous materials or large protein aggregates, has prompted the development of alternative degradation strategies [54]. Lysosome-targeting approaches have thus emerged as complementary strategies that significantly expand the druggable proteome to include membrane proteins, extracellular proteins, and protein aggregates that are inaccessible to proteasomal degradation [55] [56].
This review comprehensively compares two pioneering lysosome-targeting technologies: Lysosome-Targeting Chimeras (LYTACs) and Autophagosome-Tethering Compounds (ATTECs), examining their mechanisms, experimental applications, and relative advantages within the expanding TPD toolkit.
LYTACs: Extending Degradation to the Extracellular Realm
LYTACs represent a groundbreaking class of degraders that recruit cell-surface lysosome-trafficking receptors to direct extracellular and membrane proteins to the endosome-lysosome pathway for degradation [56]. These molecules are typically composed of three key elements: a target-binding moiety (often an antibody or small molecule), a linker region, and a ligand that binds to a lysosome-targeting receptor [57].
The most well-established LYTAC systems utilize either the asialoglycoprotein receptor (ASGPR), primarily expressed in the liver, or the more ubiquitously expressed cation-independent mannose-6-phosphate receptor (CI-M6PR) [58]. The degradation mechanism involves: (1) simultaneous engagement of the target protein and lysosome-trafficking receptor by the LYTAC molecule, (2) receptor-mediated endocytosis of the ternary complex, (3) vesicular trafficking through the endosomal system, and (4) final degradation in the lysosome [56] [57].
ATTECs: Harnessing Autophagy for Intracellular Degradation
ATTECs represent a distinct approach that directly engages the macroautophagy pathway for degrading intracellular targets [55]. First reported in 2019, these bifunctional compounds consist of a target-binding ligand connected to a LC3-binding moiety, typically through a flexible linker [55] [54].
The molecular mechanism of ATTECs operates through: (1) binding to both the target protein and LC3 (microtubule-associated protein 1A/1B-light chain 3) on the expanding phagophore membrane, (2) tethering the target to the forming autophagosome, (3) sequestration of the target within the completed autophagosome, and (4) autophagosome-lysosome fusion followed by degradation of the cargo [55]. Unlike other autophagy-based degraders, ATTECs directly engage core autophagy machinery rather than relying on intermediary receptors.
Table 1: Key Characteristics of LYTAC and ATTEC Platforms
| Feature | LYTAC | ATTEC |
|---|---|---|
| Primary Degradation Pathway | Endosome-lysosome pathway | Autophagy-lysosome pathway |
| Target Localization | Extracellular, membrane proteins | Intracellular proteins, organelles |
| Key Effector Molecule | Lysosome-trafficking receptors (e.g., CI-M6PR, ASGPR) | LC3 on phagophore membrane |
| Degradation Scope | Proteins, protein complexes | Proteins, protein aggregates, organelles |
| Cellular Process | Receptor-mediated endocytosis | Macroautophagy |
| Reported Degradation Efficiency | ~70% for membrane proteins [56] | Varies by target; demonstrated for mutant HTT [55] |
Comparative Mechanism Visualization
The diagram below illustrates the distinct cellular pathways engaged by LYTACs and ATTECs:
Experimental Approaches and Methodologies
LYTAC Implementation and Validation
LYTAC Design Considerations: LYTAC development requires careful selection of three components: (1) a high-affinity target binder (typically monoclonal antibodies or small molecules), (2) an appropriate linker chemistry, and (3) a lysosome-targeting ligand. For CI-M6PR-recruiting LYTACs, the targeting moiety is often a synthetic glycopeptide containing M6P residues, while ASGPR-directed LYTACs utilize N-acetylgalactosamine (GalNAc) ligands [56] [57].
Key Experimental Protocols:
- Target Engagement Validation: Surface plasmon resonance (SPR) or bio-layer interferometry to confirm binding affinity to both target protein and lysosomal receptor.
- Degradation Assessment: Western blotting of whole cell lysates to quantify reduction in target protein levels, typically showing 50-80% degradation within 24 hours [56].
- Pathway Specificity Controls: Co-treatment with lysosomal inhibitors (e.g., bafilomycin A1, chloroquine) but not proteasomal inhibitors (e.g., MG132) to confirm lysosomal degradation.
- Internalization Tracking: Immunofluorescence microscopy using labeled LYTACs to visualize target internalization and co-localization with lysosomal markers (LAMP1/2).
Critical Optimization Parameters:
- Linker length and composition significantly impact LYTAC efficacy, with optimal lengths typically determined empirically [59]
- Receptor expression levels vary by cell type, necessitating validation in physiologically relevant models
- Binding affinity to the lysosomal receptor must balance between efficient recruitment and potential receptor antagonism
ATTEC Implementation and Validation
ATTEC Design Considerations: ATTECs require identification of: (1) target-binding ligands with sufficient affinity and specificity, and (2) LC3-interacting compounds that bind LC3 without disrupting autophagic flux. The linker connecting these moieties must provide appropriate spatial orientation for simultaneous engagement [55].
Key Experimental Protocols:
- Ternary Complex Formation: Co-immunoprecipitation assays to confirm ATTEC-mediated association between target protein and LC3.
- Degradation Kinetics: Time-course western blotting to measure target depletion, with typical DC50 values in the nanomolar range and significant degradation observed within 4-8 hours [55].
- Pathway Specificity: Treatment with autophagy inhibitors (e.g., 3-methyladenine for early autophagy inhibition; bafilomycin A1 for lysosomal inhibition) to confirm autophagy dependence.
- Autophagic Flux Monitoring: Use of tandem fluorescent LC3 reporters (e.g., mRFP-GFP-LC3) to distinguish autophagosomes (yellow puncta) from autolysosomes (red puncta), ensuring ATTECs do not impair autophagosome-lysosome fusion.
Critical Optimization Parameters:
- LC3-binding affinity must be balanced to avoid disrupting endogenous autophagy
- ATTEC compounds should be evaluated for potential off-target binding to other LC3 family proteins
- Degradation efficiency should be correlated with autophagic flux measurements
Table 2: Experimental Comparison of LYTACs and ATTECs
| Experimental Parameter | LYTAC | ATTEC |
|---|---|---|
| Key Validation Assays | Internalization imaging, lysosomal inhibition, receptor binding | Ternary complex IP, autophagic flux assays, autophagy inhibition |
| Typical Degradation Timeline | 12-24 hours | 4-12 hours |
| Optimal Concentration Range | nM-μM (varies by target) | nM (demonstrated for multiple targets) |
| Pathway Inhibition Controls | Bafilomycin A1, chloroquine, lysosome acidification inhibitors | 3-Methyladenine, wortmannin, bafilomycin A1 |
| Critical Design Element | Receptor-binding ligand affinity and specificity | LC3-binding moiety and linker length |
| Primary Readout Methods | Western blot (membrane proteins), flow cytometry, ELISA | Western blot, immunofluorescence, protein aggregation assays |
Research Reagent Solutions
Successful implementation of LYTAC and ATTEC technologies requires specialized reagents and methodological approaches:
Table 3: Essential Research Reagents for Lysosome-Targeting Degraders
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Pathway Inhibitors | Bafilomycin A1 (lysosomal acidification), chloroquine (lysosomal function), 3-methyladenine (autophagy initiation) | Mechanism validation and pathway specificity confirmation |
| Molecular Tools | Tandem fluorescent LC3 constructs (mRFP-GFP-LC3), lysosomal markers (LAMP1/2 antibodies), ubiquitin-binding domain probes | Tracking autophagic flux and cellular localization |
| Detection Reagents | pH-sensitive dyes (LysoTracker), degradation-resistant tags (HA, FLAG), ECL substrates for western blot | Quantifying target engagement and degradation efficiency |
| Linker Chemistries | PEG-based linkers, alkyl chains, cleavable linkers (protease-sensitive) | Optimizing degrader conformation and bioavailability |
| Cell Line Models | HEK293T (transient transfection), HEPG2 (ASGPR expression), neuronal models (protein aggregation) | Evaluating cell-type specific efficacy and therapeutic potential |
Comparative Advantages and Limitations
LYTAC Strategic Considerations
Key Advantages:
- Broad target scope: Capable of degrading diverse extracellular and membrane proteins, including receptors, ligands, and circulating proteins [56]
- Tissue-specific targeting: ASGPR-directed LYTACs enable liver-specific degradation, potentially reducing off-tissue effects [57]
- Antibody compatibility: Leverages well-established antibody therapeutics as targeting moieties
Notable Limitations:
- Receptor dependence: Efficacy depends on expression levels of lysosome-trafficking receptors, varying by cell type [55]
- Endocytosis efficiency: Cellular uptake rates can limit degradation kinetics and efficacy
- Linker optimization: Requires extensive empirical optimization of linker composition and length [59]
ATTEC Strategic Considerations
Key Advantages:
- Aggregate targeting: Demonstrated efficacy in degrading protein aggregates relevant to neurodegenerative diseases [55]
- Organelle degradation: Potential to target dysfunctional organelles, expanding beyond proteostasis
- Direct pathway engagement: Bypasses ubiquitination requirements, potentially reducing off-target effects
Notable Limitations:
- Target identification: Requires development of high-affinity binders for each target of interest
- Autophagy modulation risk: Potential to interfere with basal autophagy if not properly optimized
- Limited to intracellular targets: Cannot address extracellular or membrane proteins
LYTACs and ATTECs represent complementary additions to the TPD arsenal, each exploiting distinct lysosomal pathways with characteristic advantages and limitations. LYTAC technology dramatically expands the druggable space to extracellular and membrane proteins, while ATTECs provide a direct mechanism for engaging intracellular targets, particularly those resistant to proteasomal degradation.
The choice between these technologies depends fundamentally on target localization and the specific pathological context. For extracellular targets and membrane proteins, LYTACs offer a pioneering approach, while ATTECs show particular promise for intracellular protein aggregates characteristic of neurodegenerative diseases. Both platforms face ongoing challenges in optimization, including linker design, tissue-specific delivery, and potential off-target effects.
Future directions will likely focus on developing next-generation degraders with enhanced specificity, improved pharmacokinetic properties, and conditional activation capabilities. As these technologies mature, they will undoubtedly provide powerful tools for both basic research and therapeutic development, significantly expanding our ability to target previously "undruggable" elements of the proteome.
Targeted protein degradation (TPD) has emerged as a revolutionary therapeutic strategy that moves beyond traditional occupancy-based inhibition by directing disease-causing proteins for elimination through cellular degradation machinery. While the ubiquitin-proteasome system (UPS) has been successfully harnessed by technologies like PROTACs, the autophagy-lysosome pathway offers distinct advantages for degrading larger substrates, including protein aggregates and damaged organelles, which are implicated in various challenging diseases [55] [43]. Within this landscape, autophagy-based degraders represent a rapidly advancing frontier, with AUTAC (AUTophagy-targeting Chimera) and AUTOTAC (AUTOphagy-TArgeting Chimera) emerging as two prominent technologies leveraging this cellular clearance pathway [55] [60].
The fundamental distinction between UPS and autophagy pathways lies in their substrate handling capacities. The UPS primarily degrades individual, short-lived proteins through the proteasome's narrow barrel-shaped structure, whereas autophagy specializes in eliminating bulkier cargoes, including protein aggregates, dysfunctional organelles, and intracellular pathogens, through lysosomal degradation [1]. This review comprehensively compares the molecular mechanisms, experimental applications, and therapeutic potential of AUTAC and AUTOTAC technologies, providing researchers with a structured framework for selecting appropriate platforms for specific protein degradation challenges.
Molecular Mechanisms and Key Differentiators
AUTAC Mechanism: S-Guanylation-Mediated Targeting
AUTAC molecules consist of a target-binding ligand connected to a degradation tag that recruits the autophagy machinery. Their mechanism hinges on S-guanylation, where the AUTAC molecule attaches a guanine derivative to cysteine residues on target proteins [55]. This modification creates a "kiss-and-run" signal that mimics natural autophagy targeting sequences, particularly the K63-linked polyubiquitin chains typically associated with mitochondrial autophagy [55] [60].
The S-guanylation event serves as a recognition signal for autophagy adaptor proteins, particularly p62/SQSTM1, which contains both ubiquitin-associated (UBA) domains and an LC3-interacting region (LIR) that facilitates cargo loading into developing autophagosomes [55] [1]. This mechanism enables AUTACs to degrade not only individual proteins but also larger cellular structures, including organelles and protein aggregates that resist proteasomal degradation [55].
AUTOTAC Mechanism: p62 Receptor Activation
AUTOTAC technology employs a fundamentally different approach centered on direct activation of the autophagy receptor p62/SQSTM1. The AUTOTAC structure comprises a target-binding ligand linked to a p62-binding moiety (termed ATL) that specifically engages the ZZ domain of p62 [60]. This binding event induces a conformational change that activates otherwise dormant p62, triggering its self-oligomerization through exposed PB1 domains and enhancing interaction with LC3 on autophagosomal membranes via exposed LIR domains [60].
Unlike AUTAC's "tag-and-degrade" approach, AUTOTAC functions as a direct molecular activator of the autophagy machinery, simultaneously enhancing autophagic flux while specifically directing targets into this accelerated degradation pathway [60]. This dual functionality—cargo targeting and pathway activation—enables efficient degradation of diverse substrates, including soluble proteins, multi-protein complexes, and degradation-resistant aggregates [60].
The table below summarizes the core mechanistic differences between AUTAC and AUTOTAC technologies:
Table 1: Fundamental Mechanism Comparison
| Feature | AUTAC | AUTOTAC |
|---|---|---|
| Primary Mechanism | S-guanylation of target proteins | Conformational activation of p62 |
| Key Binding Site | Target protein cysteine residues | ZZ domain of p62 |
| Ubiquitin Dependence | Utilizes ubiquitin-like signaling | Ubiquitin-independent |
| p62 Role | Adaptor for guanylated proteins | Directly activated degradation receptor |
| Autophagic Flux | Utilizes existing autophagy activity | Enhances autophagic flux |
| Substrate Scope | Proteins, organelles, aggregates | Proteins, aggregates, resistant species |
Figure 1: Comparative molecular mechanisms of AUTAC and AUTOTAC degraders. AUTAC (red pathway) functions through S-guanylation of target proteins, while AUTOTAC (blue pathway) activates p62 via ZZ domain binding, triggering oligomerization and autophagic encapsulation.
Experimental Data and Performance Comparison
Quantitative Degradation Efficiency
Direct comparison of degradation efficiency between AUTAC and AUTOTAC platforms reveals significant differences in potency and substrate scope. The following table summarizes key performance metrics reported across independent studies:
Table 2: Performance Metrics of AUTAC vs. AUTOTAC
| Parameter | AUTAC | AUTOTAC | Experimental Context |
|---|---|---|---|
| DC50 (Model Substrate) | ~100 nM (FKBP12) | ~10-100 nM (ERβ) | Soluble protein degradation [60] |
| Degradation Maximum | 70-80% | >90% | Optimal efficiency [60] |
| Time to Maximum Effect | 12-24 hours | 6-12 hours | Kinetics in cell culture [60] |
| Aggregate Clearance | Moderate | High | Neurodegenerative disease models [60] |
| Organelle Degradation | Demonstrated (mitochondria) | Limited data | Mitochondrial quality control [55] |
| Hook Effect | Present | Present | High concentration saturation [61] |
AUTOTAC platforms have demonstrated particularly robust degradation efficiency against challenging targets, including metabolic enzymes and transcriptional regulators. For instance, AUTOTACs targeting estrogen receptor beta (ERβ) achieved DC50 values in the nanomolar range (10-100 nM) with maximal degradation exceeding 90% in cellular models [60]. Similarly, AUTOTACs directed against pyruvate kinase M2 (PKM2) and methionine adenosyltransferase 2A (MAT2A) exhibited potent degradation with DC50 values of approximately 50 nM and 80 nM respectively, effectively suppressing cancer cell proliferation [60].
AUTAC molecules have shown efficacy in degrading endogenous proteins such as FKBP12 and HDAC family members, with reported DC50 values around 100 nM, though with generally lower maximal degradation efficiency (typically 70-80%) compared to AUTOTAC platforms [55] [60]. Both technologies exhibit the characteristic "hook effect" common to bivalent degraders, where degradation efficiency decreases at high concentrations due to preferential formation of binary complexes over productive ternary complexes [61].
Substrate Scope and Specificity
The autophagy-lysosome pathway offers a broader substrate scope compared to UPS-based degradation systems, with both AUTAC and AUTOTAC capable of degrading proteins, protein aggregates, and organelles that resist proteasomal degradation [55] [60]. However, important distinctions exist in their specific applications:
AUTAC Substrate Range: AUTAC technology has demonstrated effectiveness in degrading fragmented mitochondria through its S-guanylation mechanism, connecting it to natural mitochondrial quality control pathways [55]. Additionally, AUTACs have successfully targeted metabolically significant proteins including FKBP12, HDAC proteins, and various transcriptional regulators, though with variable efficiency across target classes [55] [60].
AUTOTAC Substrate Range: AUTOTAC platforms exhibit remarkable versatility, efficiently degrading diverse substrate types including soluble oncoproteins (e.g., ERβ, BRD4), metabolic enzymes (e.g., PKM2), and neurodegenerative disease-associated aggregates (e.g., mutant Huntingtin, Tau) [60]. The p62 activation mechanism appears particularly effective against aggregation-prone proteins that typically resist proteasomal degradation, with studies demonstrating clearance of intracellular aggregates implicated in proteinopathies [60].
Table 3: Substrate Scope and Specificity Comparison
| Substrate Category | AUTAC Examples | AUTOTAC Examples | Relative Efficiency |
|---|---|---|---|
| Soluble Oncoproteins | METAP2, HDACs | ERβ, BRD4, PKM2 | AUTOTAC > AUTAC |
| Protein Aggregates | Limited data | Huntingtin, Tau | AUTOTAC demonstrated |
| Organelles | Mitochondria | Limited data | AUTAC demonstrated |
| Signaling Proteins | FKBP12, Transcriptional regulators | MAT2A, STING | Comparable |
| Membrane Proteins | Limited data | Limited data | Both limited |
Experimental Protocols and Methodologies
AUTAC Degradation Assay Protocol
Cell Culture and Treatment:
- Seed appropriate cell lines (e.g., HEK293, HeLa, or primary cells) in 6-well or 12-well plates and culture until 60-70% confluence
- Prepare serial dilutions of AUTAC compounds in DMSO (typically 1 nM to 10 μM final concentration)
- Treat cells for predetermined durations (usually 6-24 hours) with appropriate vehicle controls
- For autophagy inhibition controls, co-treat with 3-methyladenine (5 mM) or bafilomycin A1 (100 nM)
Sample Collection and Analysis:
- Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors
- Quantify protein concentration using BCA assay and normalize samples
- Perform Western blotting with antibodies against target protein and loading controls (e.g., GAPDH, β-actin)
- Quantify band intensity using densitometry software (ImageJ or similar)
- Calculate degradation efficiency as percentage reduction relative to vehicle control
- Determine DC50 values using nonlinear regression analysis (GraphPad Prism or equivalent)
Validation Experiments:
- Confirm autophagy dependence through co-treatment with autophagy inhibitors
- Assess specificity through proteomic analysis or examination of unrelated proteins
- Visualize subcellular localization via immunofluorescence microscopy with organelle markers
AUTOTAC Degradation Assay Protocol
Cell Culture and Treatment:
- Culture cells in appropriate media to 50-60% confluence in multi-well plates or chamber slides
- Prepare AUTOTAC compounds in DMSO (typically 10 nM to 1 μM concentration range)
- Treat cells for 6-48 hours depending on target protein half-life
- Include controls: DMSO vehicle, p62 knockdown cells, and autophagy inhibitors (chloroquine 50 μM or bafilomycin A1 100 nM)
Degradation Efficiency Assessment:
- For Western blot analysis: Harvest cells, lyse, and process as described for AUTAC protocol
- For immunofluorescence: Fix cells with 4% PFA, permeabilize with 0.1% Triton X-100, block with 5% BSA, and incubate with primary antibodies (target protein, p62, LC3)
- Image using confocal microscopy and quantify puncta formation and co-localization
- For flow cytometry: Use GFP-tagged targets or antibody staining to quantify protein levels in individual cells
p62 Activation and Oligomerization Assays:
- Perform native PAGE to detect p62 oligomerization
- Conduct co-immunoprecipitation to assess p62-LC3 interaction
- Measure autophagic flux using LC3 turnover assays (LC3-II accumulation in presence/absence of lysosomal inhibitors)
Figure 2: Generalized experimental workflow for evaluating autophagy-based degraders. The protocol encompasses cell treatment, degradation efficiency measurement, and mechanistic validation, applicable to both AUTAC and AUTOTAC platforms with specific modifications.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of AUTAC and AUTOTAC research requires specific reagents and tools to design, validate, and optimize degradation experiments. The following table outlines essential components for establishing these platforms in research settings:
Table 4: Essential Research Reagents for Autophagy-Based Degradation Studies
| Reagent Category | Specific Examples | Function/Application | Availability |
|---|---|---|---|
| Target Binders | PHTPP (ERβ ligand), JQ1 (BRD4 ligand), FKBP12 ligands | Target protein recognition modules | Commercial suppliers |
| Autophagy Modulators | YOK-2204, YOK-1304, YTK-105 (p62 ligands) | p62 binding and activation (AUTOTAC) | Specialty suppliers [60] |
| Linker Chemistry | PEG chains, alkyl chains, custom spacers | Connect target binders to degradation tags | Chemical synthesis |
| Validation Antibodies | Anti-p62, anti-LC3, anti-ubiquitin, target-specific antibodies | Mechanism confirmation and efficiency measurement | Multiple suppliers |
| Autophagy Inhibitors | Bafilomycin A1, chloroquine, 3-methyladenine | Pathway inhibition controls | Commercial suppliers |
| Cell Lines | HEK293, HeLa, neuronal models, cancer lines | Degradation efficiency screening | Cell repositories |
| Reporters | GFP-LC3, RFP-GFP-LC3, ubiquitin sensors | Autophagic flux measurement | Addgene, commercial |
For AUTOTAC platforms specifically, critical reagents include validated p62-ZZ domain ligands (YOK-2204, YOK-1304, YTK-105) that serve as the autophagy-targeting ligands (ATLs) [60]. These compounds have demonstrated specific binding to the p62 ZZ domain with dissociation constants in the micromolar range, effectively inducing p62 oligomerization and activation of the autophagy machinery [60].
Additionally, researchers should employ p62 knockout cell lines and siRNA-mediated p62 knockdown to confirm mechanism specificity, alongside standard autophagy reporter systems (e.g., GFP-LC3, RFP-GFP-LC3) to monitor autophagic flux enhancement in response to degrader treatment [55] [60].
AUTAC and AUTOTAC technologies represent distinct approaches to harnessing autophagy for targeted protein degradation, each with characteristic strengths and optimal applications. AUTAC's S-guanylation mechanism connects to natural quality control pathways, showing particular promise for organelle degradation and targets with accessible cysteine residues. AUTOTAC's direct activation of p62 provides robust degradation efficiency against diverse substrates, including challenging aggregation-prone proteins, while simultaneously enhancing cellular autophagic capacity [55] [60].
The emerging paradigm in targeted degradation emphasizes contextual deployment of complementary technologies based on specific research or therapeutic objectives. AUTAC technology may be preferable for mitochondrial quality control applications and situations where endogenous p62 levels are limiting, while AUTOTAC platforms offer advantages for aggressive aggregate clearance and targets requiring maximal degradation efficiency [60]. Both platforms significantly expand the druggable proteome beyond UPS-based approaches, particularly for neurodegenerative disease targets characterized by protein aggregation [55] [60].
Future directions include developing conditional activation systems, tissue-selective targeting approaches, and combination therapies that leverage synergistic effects between autophagy-based degradation and complementary therapeutic modalities [61] [43]. As these technologies mature, they will undoubtedly provide researchers with increasingly precise tools to interrogate biological systems and address previously intractable disease targets through targeted protein degradation.
Leveraging Chaperone-Mediated Autophagy (CMA) for Targeted Degradation
Maintaining cellular protein homeostasis, or proteostasis, is absolutely essential for proper cellular functioning and survival. Two major systems mediate the complete degradation of intracellular proteins into their constitutive amino acids: the Ubiquitin-Proteasome System (UPS) and the autophagy-lysosomal system [62] [10]. The UPS is a multi-subunit protease complex in the cytosol which permits entry and subsequent degradation of proteins tagged with one or more covalently bound ubiquitin molecules. Most proteasome substrates are short-half-life proteins such as newly synthesized, misfolded, and critical regulatory proteins involved in cell division, signaling, and transcription [62] [10]. In contrast, the autophagic/lysosomal system is dedicated to degradation of both intracellular and extracellular components, with three main types identified in mammals: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [62] [63]. This guide provides a comprehensive comparison of these pathways, with special emphasis on leveraging CMA for targeted degradation, including experimental methodologies, key reagents, and therapeutic applications.
Molecular Mechanisms of CMA
The Stepwise Process of CMA
Chaperone-mediated autophagy is a uniquely selective form of autophagy by which specific cytosolic proteins are transported one-by-one across the lysosomal membrane for degradation [62]. The process involves several distinct steps:
Substrate Recognition: Cytosolic chaperones, predominantly heat shock cognate protein 70 (HSC70), recognize and bind to proteins containing a KFERQ-like pentapeptide motif [62] [63]. It is estimated that approximately 30% of cytosolic proteins bear this targeting sequence [62].
Lysosomal Binding: The chaperone-substrate complex is directed to the lysosomal membrane where it binds to the cytosolic tail of lysosome-associated membrane protein type 2A (LAMP-2A), which acts as the CMA receptor [64] [63].
Protein Translocation: The substrate protein unfolds and translocates across the lysosomal membrane through a multimeric LAMP-2A translocation complex. A lumenal form of HSC70 (lys-HSC70) inside lysosomes completes substrate translocation [62].
Degradation and Recycling: The translocated protein is rapidly degraded by lysosomal hydrolases, and the resulting amino acids are recycled back to the cytosol [63].
Regulatory Mechanisms
CMA activity is primarily regulated by changes in the composition of the lysosomal membrane and by the levels of LAMP-2A, which serves as the rate-limiting step in CMA [62] [65]. newly discovered regulatory pathways include NRF2 and p38-TFEB signaling, acting as positive and negative regulatory pathways of CMA, respectively [63].
Comparative Analysis of Protein Degradation Pathways
The following table provides a systematic comparison of the major protein degradation pathways in mammalian cells:
Table 1: Comparative Analysis of Protein Degradation Pathways
| Feature | Ubiquitin-Proteasome System (UPS) | Macroautophagy | Chaperone-Mediated Autophagy (CMA) |
|---|---|---|---|
| Degradation Mechanism | ATP-dependent degradation by 26S proteasome | Bulk sequestration via autophagosomes that fuse with lysosomes | Selective translocation of unfolded proteins across lysosomal membrane |
| Specificity | Selective (via ubiquitin tagging) | Non-selective (bulk) or selective (via receptors) | Highly selective (KFERQ motif requirement) |
| Key Components | E1-E3 enzymes, proteasome | ATG proteins, lysosomal enzymes | HSC70, LAMP-2A, lysosomal HSC70 |
| Substrate Types | Short-lived, regulatory, misfolded proteins | Organelles, protein aggregates, pathogens | Soluble cytosolic proteins with KFERQ motif |
| Energy Requirements | ATP-dependent | ATP-dependent | ATP-dependent |
| Physiological Roles | Protein quality control, cell cycle regulation, signaling | Nutrient recycling during starvation, organelle turnover | Metabolic adaptation, stress response, glucose/lipid regulation |
| Response to Stress | Activated by various stresses | Strongly induced by starvation | Activated by oxidative stress, hypoxia, toxin exposure |
Table 2: Quantitative Comparison of Degradation Efficiency
| Parameter | UPS | CMA | Experimental Evidence |
|---|---|---|---|
| Degradation Rate | Minutes to hours | Hours to days | MG132 inhibition studies show PGC1α half-life differences [64] |
| Motif Recognition | Ubiquitin tag (various linkages) | KFERQ-like motif (30% of proteome) | KFERQ finder analysis; mutant studies [64] [62] |
| Substrate Size | Limited by proteasome pore size | Unlimited (unfolding required) | PGC1α (90 kDa) successfully degraded via CMA [64] |
| Tissue Distribution | Ubiquitous | Highest in liver, kidney, heart | Tissue-specific LAMP2A expression patterns [65] [63] |
Experimental Approaches for Studying CMA
Methodologies for Monitoring CMA Activity
Several well-established experimental approaches enable researchers to monitor CMA activity and substrate degradation:
CMA Reporter Assays: The most direct method involves monitoring the translocation of CMA substrates into lysosomes. The classic assay uses radiolabeled substrates incubated with isolated lysosomes, followed by quantification of degraded substrates [62]. Modern adaptations employ fluorescent-tagged CMA substrates.
Lysosomal Isolation and Analysis: Isolation of lysosomes from tissues or cells via density gradient centrifugation allows for direct measurement of LAMP-2A levels, LAMP-2A multimerization status, and association of HSC70 with lysosomal membranes [64] [62].
Immunoblotting of CMA Components: Western blot analysis of LAMP-2A protein levels provides an indirect measure of CMA capacity, though activity should be confirmed functionally [64] [65].
CMA Activity Sensors: Genetically encoded reporters containing canonical KFERQ motifs fused to fluorescent proteins allow real-time monitoring of CMA activity in live cells [64].
Experimental Workflow for CMA Substrate Validation
The following diagram illustrates a validated experimental workflow for confirming whether a protein of interest is a bona fide CMA substrate:
Key Experimental Models for CMA Research
In Vitro Systems:
In Vivo Models:
Research Reagent Solutions
The following table compiles essential research reagents for studying CMA and their specific applications:
Table 3: Essential Research Reagents for CMA Studies
| Reagent Category | Specific Examples | Function/Application | Experimental Evidence |
|---|---|---|---|
| CMA Activators | AR7, QX77 | Enhance CMA activity by stabilizing LAMP-2A multimers | AR7 ameliorated disc degeneration in rat IDD model [65] |
| CMA Inhibitors | ATRA (All-trans retinoic acid) | Suppresses CMA activity by reducing LAMP-2A levels | ATRA increased senescence markers in NPC [65] |
| Genetic Tools | shLamp2a, LAMP2A OE lentivirus, CRISPR/Cas9 LAMP2A KO | Modulate CMA capacity for functional studies | BAT-specific Lamp2a knockdown improved energy metabolism [64] |
| Detection Antibodies | Anti-LAMP2A, Anti-HSC70, Anti-PGC1α | Quantify protein levels and interactions | Co-IP showed HSC70-LAMP2A interaction enhanced by thermal stress [64] |
| CMA Reporters | KFERQ-GFP constructs, KFERQ-mutant controls | Monitor substrate degradation and CMA activity | KFERQ-mutant PGC1α resistant to degradation [64] |
| Lysosomal Markers | LysoTracker, LAMP1 antibodies | Identify and isolate lysosomal compartments | Used in lysosomal isolation for CMA activity assays [64] [62] |
CMA in Cellular Physiology and Disease
Physiological Roles of CMA
CMA plays essential roles in diverse physiological processes through its selective degradation of key regulatory proteins:
Metabolic Regulation: CMA degrades key enzymes related to carbohydrate and lipid metabolism, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH), glycerol-3-phosphate dehydrogenase 2, and acyl-coenzyme A dehydrogenase long chain [63].
Thermal Stress Response: Under thermal stress, CMA targets thermogenic proteins like PGC1α for degradation, contributing to brown adipose tissue whitening at thermoneutral conditions [64].
Immune Function: CMA maintains T cell activation by degrading negative regulators such as Itch and regulator of calcineurin-1 (Rcan-1) [63].
Cell Cycle Control: CMA helps resist hypoxia-mediated cell cycle arrest by degrading hypoxia-inducible factor-1 alpha (HIF-1α) [63].
CMA Dysregulation in Disease
The following diagram illustrates the consequences of CMA dysfunction across different pathological conditions:
Neurodegenerative Diseases: CMA dysfunction contributes to Parkinson's disease, Alzheimer's disease, and other neurodegenerative disorders through impaired clearance of aggregation-prone proteins like α-synuclein and tau [63].
Metabolic Diseases: Impaired CMA disrupts glucose and lipid homeostasis, contributing to conditions like diabetes and obesity [64] [63].
Cancer: CMA plays dual roles in tumorigenesis, with both tumor-suppressive and tumor-promoting activities depending on context [63] [66].
Aging: CMA activity declines with age across multiple tissues, contributing to the accumulation of damaged proteins and cellular dysfunction [65] [63].
Intervertebral Disc Degeneration (IDD): CMA impairment promotes cellular senescence in nucleus pulposus cells through DYRK1A accumulation, driving disc degeneration [65].
Therapeutic Targeting of CMA
The strategic manipulation of CMA holds significant therapeutic potential for various diseases. Both CMA activation and inhibition represent viable approaches depending on the pathological context:
CMA Activation Strategies:
CMA Inhibition Approaches:
Recent advances in targeted protein degradation technologies, including PROTACs and LYTACs, highlight the potential for designing synthetic substrates that harness CMA for specific protein elimination. The selectivity of CMA for KFERQ-containing proteins provides a natural framework for engineering degraders that mimic this motif to direct pathological proteins to lysosomal degradation.
CMA represents a highly selective protein degradation pathway with distinct advantages and limitations compared to UPS and macroautophagy. Its singular selectivity for KFERQ-containing proteins, precise regulation at the level of substrate translocation, and involvement in diverse physiological processes make it an attractive target for therapeutic intervention. While technical challenges remain in specifically modulating CMA activity without affecting parallel degradation pathways, recent advances in understanding CMA molecular mechanisms and regulatory networks have created unprecedented opportunities for leveraging this pathway in targeted protein degradation strategies. The continued development of more specific CMA modulators and engineered substrate constructs will further enhance our ability to harness this natural degradation system for research and therapeutic applications.
Targeted protein degradation (TPD) represents a revolutionary therapeutic strategy that moves beyond traditional inhibition to achieve the complete elimination of disease-causing proteins. [67] This approach leverages the cell's intrinsic proteolytic systems—primarily the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system (ALS)—to selectively degrade pathological proteins. [67] [68] However, the efficacy of molecular degraders like proteolysis-targeting chimeras (PROTACs) and autophagy-targeting chimeras (AUTACs) is often constrained by challenges including poor cellular permeability, limited solubility, and unpredictable structure-activity relationships. [69] Nanotechnology has emerged as a powerful enabling platform to overcome these limitations, giving rise to nanoparticle-mediated protein degraders (NanoPDs) that enhance delivery, improve bioavailability, and expand the therapeutic potential of TPD technologies. [69] This review provides a comprehensive comparison of how nanotechnology interfaces with both major cellular degradation pathways—UPS and autophagy—to advance the field of targeted degradation, with specific focus on experimental approaches and performance metrics relevant to drug development professionals.
Cellular Degradation Pathways: UPS versus Autophagy
The Ubiquitin-Proteasome System (UPS)
The UPS is a primary proteolytic pathway responsible for degrading short-lived, misfolded, and damaged proteins. [10] [70] This ATP-dependent process involves a coordinated enzymatic cascade where E1 (activating), E2 (conjugating), and E3 (ligating) enzymes conjugate ubiquitin chains to target proteins. [10] Polyubiquitinated proteins are then recognized and degraded by the 26S proteasome, a multi-subunit complex comprising a 20S catalytic core and 19S regulatory particles. [70] The K48-linked ubiquitin chains serve as the classical signal for proteasomal degradation. [70] Key UPS-based TPD technologies include PROTACs and molecular glues, which redirect E3 ubiquitin ligases to target proteins of interest for ubiquitination and subsequent proteasomal degradation. [67] [69]
The Autophagy-Lysosome System (ALS)
Autophagy is a highly conserved catabolic process that degrades long-lived proteins, protein aggregates, and damaged organelles through lysosomal enzymes. [55] [68] This pathway involves the formation of double-membrane vesicles called autophagosomes that engulf cytoplasmic cargo, which then fuse with lysosomes to form autophagolysosomes where degradation occurs. [55] [11] Core autophagy-related (ATG) proteins regulate this process, with LC3/ATG8 serving as a specific marker for autophagosome formation. [55] Unlike the UPS, autophagy can handle bulkier substrates including protein aggregates and organelles, making it particularly valuable for addressing pathological conditions like neurodegenerative diseases. [55] [68] Autophagy-based TPD strategies include AUTACs, AUTOTACs, and ATTECs, which leverage selective autophagy receptors like p62/SQSTM1 to bridge target proteins to the autophagic machinery. [55] [68]
Table 1: Comparative Features of UPS and Autophagy Pathways
| Parameter | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome System (ALS) |
|---|---|---|
| Primary Substrates | Short-lived, soluble proteins [68] | Long-lived proteins, aggregates, organelles [68] |
| Degradation Machinery | 26S proteasome [70] | Lysosomal hydrolases [68] |
| Key Recognition Signal | K48-linked ubiquitin chains [70] | Multiple ubiquitin linkages (K63, K48, etc.) [70] |
| Energy Requirement | ATP-dependent [10] | ATP-dependent [11] |
| TPD Technologies | PROTACs, Molecular Glues [67] [69] | AUTACs, ATTECs, AUTOTACs [55] [68] |
| Nanoparticle Entry | Cytosolic delivery required [69] | Lysosomal delivery advantageous [69] |
Pathway Interdependence and the Role of p62
The UPS and autophagy are interconnected systems that maintain proteostasis. The multifunctional protein p62/SQSTM1 serves as a critical link between these pathways, functioning as a selective autophagy receptor that delivers ubiquitinated cargo to autophagosomes via its LC3-interacting region (LIR). [70] Simultaneously, p62 can shuttle ubiquitinated proteins to the proteasome through its PB1 domain. [70] This dual functionality enables cross-talk between degradation pathways, particularly under proteotoxic stress where inhibition of one system can compensatory activate the other. [70]
Diagram 1: Cellular protein degradation pathways and nanoparticle interface. Nanoparticle-mediated protein degraders (NanoPDs) can enhance delivery to both ubiquitin-proteasome and autophagy-lysosome systems, leveraging the p62 adaptor that bridges these pathways.
Nanotechnology Platforms for Targeted Protein Degradation
Nanoparticle-Mediated Protein Degraders (NanoPDs)
Nanoparticle-mediated protein degraders (NanoPDs) represent a diverse class of nanoscale materials engineered to facilitate targeted protein degradation through various mechanisms. First reported approximately 17 years ago, NanoPDs have evolved to encompass a wide range of materials including lipid-based nanoparticles, gold nanoparticles, polymeric nanoparticles, and DNA nanostructures. [69] These platforms address fundamental limitations of conventional degraders by improving solubility, enhancing cellular uptake, and protecting payloads from premature degradation. [69] The design principles governing NanoPDs focus on optimizing the nano-bio interface to achieve efficient intracellular delivery while minimizing nonspecific interactions.
Mechanism of Action and Material Considerations
NanoPDs operate through multiple mechanisms depending on their composition and target pathway. For UPS-targeting degraders, nanoparticles often serve as delivery vehicles for PROTACs or molecular glues, facilitating cytosolic release to enable ternary complex formation between target protein and E3 ligase. [69] For autophagy-mediated degradation, nanoparticles can directly engage autophagic machinery through surface functionalization with LC3-binding peptides or autophagy receptors. [55] [69] Material selection critically influences degradation efficacy—lipid nanoparticles excel in encapsulating hydrophobic degraders, gold nanoparticles enable precise surface engineering for multivalent interactions, and polymeric nanoparticles offer controlled release kinetics. [69] Size optimization is crucial, as demonstrated by early studies showing 40-60 nm gold nanoparticles achieving maximal HER2 receptor degradation. [69]
Table 2: Comparison of NanoPD Platforms for UPS versus Autophagy Pathways
| NanoPD Platform | Mechanism of Action | Advantages | Limitations | Experimental Efficacy |
|---|---|---|---|---|
| Lipid Nanoparticles (LNP) | PROTAC/molecular glue delivery; Endosomal escape [69] | High biocompatibility; Clinical translation feasibility [69] | Lysosomal entrapment; Limited tissue targeting [69] | DiD-LNP: 106.7 nm size, 0.145 PDI [71] |
| Gold Nanoparticles | Surface conjugation with protein binders; Multivalent engagement [69] | Precise surface engineering; Tunable size & shape [69] | Potential cytotoxicity; Long-term accumulation concerns [69] | HER2 degradation: 40-60 nm optimal [69] |
| Polymeric Nanoparticles | Controlled release of degraders; Stabilization of ternary complexes [69] | Tunable degradation kinetics; Versatile chemistry [69] | Complexity in manufacturing; Batch-to-batch variability [69] | Enhanced cellular accumulation in G2/M phase [71] |
| DNA Nanostructures | Precise spatial organization of protein binders [69] | Molecular precision; Biodegradability [69] | Limited payload capacity; Stability in biological fluids [69] | Emerging technology with promising in vitro data [69] |
Experimental Approaches and Methodologies
Assessing Cellular Accumulation and Degradation Efficacy
Evaluating NanoPD efficacy requires sophisticated methodologies that account for cell cycle dependencies and autophagy status. Advanced platforms integrate multiple technical approaches including PIP-FUCCI transfection for cell cycle phase distinction, ATG7 knockout to specifically block autophagy, and 3D reconstruction to stereoscopically evaluate nanoparticle accumulation. [71] Experimental protocols typically involve:
- Cell Synchronization and Phase Separation: Using mitotic shake-off to separate cell cycle phases without chemical synchronizers that might disrupt cellular physiology. [71]
- Autophagy Modulation: Employing isogenic wild-type and ATG7 knockout cell lines to eliminate confounding effects of pharmacological autophagy inhibitors. [71]
- Multidimensional Imaging: Combining high-resolution confocal laser scanning microscopy with 3D reconstruction to account for cell geometric characteristics and heterogeneous intracellular nanoparticle distribution. [71]
Quantitative measurements include total fluorescence intensity (TFI) and mean fluorescence intensity (MFI) at both 2D and 3D levels, providing comprehensive assessment of nanoparticle accumulation across different cellular conditions. [71]
Experimental Workflow for NanoPD Evaluation
Diagram 2: Experimental workflow for evaluating NanoPD efficacy. The protocol encompasses nanoparticle synthesis, thorough characterization, cell model preparation with autophagy and cell cycle indicators, treatment, and multi-modal analysis to assess degradation efficiency.
Key Research Reagents and Experimental Tools
Table 3: Essential Research Reagents for NanoPD Development and Evaluation
| Reagent/Cell Line | Function/Application | Key Characteristics | Experimental Utility |
|---|---|---|---|
| PIP-FUCCI Plasmid | Cell cycle phase indicator [71] | Fluorescent labeling of G1 (red), S (yellow), G2 (green) phases [71] | Distinguishes cell cycle-dependent NanoPD accumulation without synchronization artifacts [71] |
| ATG7 KO Cell Lines | Autophagy-deficient models [71] | Specific blockade of autophagosome formation [71] | Investigates autophagy-specific degradation mechanisms without pharmacological inhibitors [71] |
| DiD-LIP/DiD-LNP | Fluorescent nanoparticle tracers [71] | Z-average: ~105 nm, PDI: <0.15, stable in serum [71] | Enables tracking of cellular accumulation and distribution of lipid-based NanoPDs [71] |
| LC3-II Antibody | Autophagosome marker detection [55] | Recognizes lipidated LC3 form on autophagosomes [55] | Measures autophagic flux in response to NanoPD treatment [55] |
| p62/SQSTM1 Antibody | Autophagy adapter protein detection [70] | Binds both ubiquitin and LC3-II [70] | Dual indicator of autophagic flux and ubiquitinated protein clearance [70] |
Performance Comparison: Nano-Enhanced UPS versus Autophagy Strategies
Degradation Efficiency and Specificity
Nanotechnology enhancement confers distinct advantages to both UPS and autophagy-based degradation approaches. For UPS-targeting strategies, NanoPDs significantly improve the cellular delivery of PROTACs, which traditionally suffer from poor membrane permeability due to their large molecular weights and complex structures. [67] [69] Lipid-based NanoPDs encapsulating PROTACs have demonstrated enhanced degradation efficiency of oncoproteins like androgen and estrogen receptors in preclinical models. [69] For autophagy-based strategies, nanoparticles functionalized with LC3-binding motifs or autophagy receptors achieve selective degradation of protein aggregates and organelles, targets that are typically refractory to UPS-mediated degradation. [55] [69] The table below summarizes key performance metrics for both approaches:
Table 4: Performance Comparison of Nano-Enhanced UPS versus Autophagy Strategies
| Performance Metric | Nano-UPS Platforms | Nano-Autophagy Platforms |
|---|---|---|
| Target Range | Soluble proteins, transcription factors [67] | Protein aggregates, organelles, pathogens [55] |
| Degradation Kinetics | Rapid (hours) [67] | Slower (hours to days) [55] |
| Catalytic Efficiency | High (substoichiometric) [69] | Moderate to high [55] |
| Tissue Penetration | Enhanced via nanocarriers [69] | Enhanced via nanocarriers [69] |
| BBB Crossability | Formulation-dependent [72] | Formulation-dependent [72] |
| Therapeutic Index | Improved with targeted nanoparticles [69] | Improved with targeted nanoparticles [69] |
| Clinical Translation | Phase III trials (ARV-471) [67] | Preclinical development [55] |
Technical Challenges and Optimization Strategies
Both NanoPD approaches face significant technical hurdles. For UPS-focused strategies, the hook effect—where high degrader concentrations disrupt ternary complex formation—persists despite nanoformulation. [67] Additionally, lysosomal entrapment of nanoparticles can sequester PROTACs away from their cytosolic targets, reducing efficacy. [69] For autophagy-focused strategies, controlling the specificity of autophagic engulfment remains challenging, with potential for off-target degradation of cellular components. [55] Protein corona formation on nanoparticles in biological fluids can also mask targeting ligands, reducing degradation specificity for both approaches. [69]
Optimization strategies include:
- Surface functionalization with cell-penetrating peptides to enhance cytosolic delivery
- Stimulus-responsive linkers that release payloads in response to intracellular cues
- Size optimization to maximize cellular uptake while minimizing clearance
- Covalent conjugation of degraders to nanoparticles to prevent premature dissociation
The integration of nanotechnology with targeted protein degradation has created powerful synergies that address fundamental limitations of both UPS and autophagy-based degradation strategies. NanoPD platforms enhance the delivery, efficacy, and specificity of molecular degraders while expanding the targetable proteome to include previously "undruggable" proteins. As the field advances, key areas for future development include the design of pathway-specific nanocarriers optimized for either UPS or autophagy engagement, the engineering of smart nanoparticles with stimulus-responsive degradation activation, and the exploration of combination approaches that simultaneously harness both degradation pathways. The ongoing clinical evaluation of PROTACs and the rapid preclinical advancement of autophagy-targeting degraders suggest that nano-enhanced TPD strategies will play an increasingly prominent role in the next generation of therapeutic modalities for cancer, neurodegenerative disorders, and other proteinopathies.
Navigating Challenges: System Impairment, Crosstalk, and Compensatory Mechanisms
In eukaryotic cells, the ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway constitute the two major pillars of protein quality control, responsible for maintaining proteostasis by eliminating misfolded, damaged, or redundant proteins [73] [2]. The UPS primarily degrades short-lived soluble proteins through a highly specific mechanism involving ubiquitination and proteasomal proteolysis [2] [36]. In contrast, autophagy handles the bulk degradation of long-lived proteins, insoluble protein aggregates, and damaged organelles through the lysosomal system [74] [2]. While these systems often function cooperatively, each has distinct vulnerabilities that can lead to catastrophic failure under pathological conditions. Protein aggregation diseases, including neurodegenerative disorders and certain cardiomyopathies, are characterized by the accumulation of toxic protein aggregates that directly impair these degradation systems [74]. This review systematically compares how protein aggregates inhibit UPS function and how blocks in autophagic flux disrupt clearance mechanisms, providing experimental approaches for investigating these critical failure points in cellular proteostasis.
UPS Inhibition by Protein Aggregates
Mechanisms of Proteasomal Inhibition
The ubiquitin-proteasome system exhibits particular vulnerability to structured protein aggregates due to the physical architecture of the proteasome itself. The 26S proteasome features a barrel-shaped 20S core particle with a narrow entrance pore that restricts access to the proteolytic chamber [1] [2]. This structure, while ideal for processing unfolded polypeptide chains, presents an insurmountable physical barrier to aggregated proteins, which exceed the steric limitations of the proteasomal gated channel [74] [1].
Key mechanisms of UPS inhibition by protein aggregates include:
Steric Hindrance: Insoluble protein aggregates and large oligomeric species cannot enter the narrow proteasomal degradation chamber, effectively resisting proteasomal degradation despite being ubiquitinated [74]. Studies demonstrate that while soluble poly-Q expansion proteins can be degraded by the proteasome, their aggregated forms remain resistant [74].
Proteasome Sequestration: Protein aggregates can actively sequester proteasomal components, effectively depleting functional proteasomes from the cellular pool. Research shows that p62/SQSTM1 aggregates can associate with inactive proteasomes, ubiquitinated proteins, and autophagosomes, suggesting a mechanism of proteasome entrapment within protein aggregates [75].
Active Inhibition: Certain protein aggregates, particularly oligomeric species, can directly inhibit proteasomal activity. In prion neuropathology models, prion oligomers specifically inhibit the catalytic β subunit activity of the proteasome, an effect that can be reversed by anti-oligomer antibodies [74].
Competitive Binding: Aggregates may monopolize ubiquitin-binding receptors such as RPN10 and RPN13 in the 19S regulatory particle, preventing other ubiquitinated substrates from accessing the degradation machinery [76].
Table 1: Experimental Evidence for UPS Inhibition by Protein Aggregates
| Aggregate Type | Experimental System | Key Findings | Citation |
|---|---|---|---|
| Poly-Q expansions | Cell models | Soluble forms degraded by UPS; aggregated forms resist degradation | [74] |
| α-synuclein | Neuronal cells | Aggregated forms inhibit proteasome function; autophagy preferentially clears aggregates | [74] |
| Prion oligomers | Prion disease models | Oligomers inhibit catalytic β subunit of proteasome; reversible with anti-oligomer antibodies | [74] |
| p62/SQSTM1 aggregates | SK-N-SH cells, MEFs | p62 aggregates contain inactive proteasomes, ubiquitinated proteins, and autophagosomes | [75] |
Experimental Assessment of UPS Function
Multiple experimental approaches have been developed to assess UPS function in the context of protein aggregation. The following protocol represents a standardized methodology for evaluating proteasomal inhibition:
Protocol 1: Assessing UPS Dysfunction in Protein Aggregation Models
Cell Treatment: Expose experimental systems (cell lines, primary neurons) to proteasome inhibitors (e.g., 1-10 μM epoxomicin or MG132) for 4-24 hours as a positive control for UPS impairment [75].
Ubiquitinated Protein Detection:
- Harvest cells and prepare lysates in RIPA buffer with proteasome inhibitors
- Perform western blot analysis using anti-ubiquitin antibodies (e.g., FK2 antibody)
- Quantify high-molecular-weight ubiquitin conjugates
Proteasome Activity Assays:
- Use fluorogenic substrates (LLVY-AMC for chymotrypsin-like, LLR-AMC for trypsin-like, LLE-AMC for caspase-like activity)
- Measure fluorescence release (excitation/emission: 380/460 nm) in cell lysates or purified proteasomes
- Normalize activities to protein content
Proteasome Localization Studies:
- Perform immunofluorescence co-staining of proteasome subunits (20S core or 19S cap) with aggregate markers (ubiquitin, p62)
- Analyze co-localization using confocal microscopy and image quantification software
Critical interpretation note: Studies consistently show that genetic or pharmacologic inhibition of autophagy alone fails to significantly increase ubiquitinated protein levels unless proteasome function is concurrently affected [75]. This underscores the hierarchical relationship between these degradation systems, with the UPS serving as the primary defense against protein aggregation.
Autophagic Flux Blockage
Mechanisms of Impaired Autophagic Clearance
While autophagy activation serves as a compensatory mechanism when UPS function is compromised, the autophagic pathway itself is susceptible to multiple points of failure. Autophagic flux represents the dynamic process encompassing autophagosome formation, cargo loading, lysosomal fusion, and content degradation. Blockage at any of these stages can disrupt protein aggregate clearance and contribute to proteostatic failure [77].
Major failure points in autophagic flux include:
Impaired Autophagosome Formation: Disruption of the ULK1 complex or ATG conjugation systems can prevent proper autophagosome initiation and elongation. Key regulators include mTORC1, which phosphorylates and inhibits ULK1 under nutrient-rich conditions [2].
Defective Cargo Recognition: In selective autophagy, adaptor proteins like p62/SQSTM1 and NBR1 link ubiquitinated cargo to LC3 on forming autophagosomes. Mutations or functional impairment of these adaptors can prevent specific targeting of protein aggregates for autophagic degradation [1].
Failed Lysosomal Fusion: Autophagosomes must fuse with lysosomes to form autolysosomes where degradation occurs. Disruption of fusion machinery (SNARE proteins, Rab GTPases) or lysosomal positioning can prevent this critical step [2].
Lysosomal Dysfunction: Reduced lysosomal acidification, depletion of hydrolytic enzymes, or impaired lysosomal biogenesis can render autophagic degradation ineffective even with successful autophagosome formation and fusion [77].
The transcription factor EB (TFEB) serves as a master regulator of lysosomal biogenesis and autophagic flux. Proteasome impairment has been shown to promote TFEB dephosphorylation and nuclear translocation, increasing expression of autophagic and lysosomal genes as a compensatory mechanism [77].
Table 2: Experimental Assessment of Autophagic Flux Blockage Points
| Flux Stage | Assessment Method | Key Markers | Interpretation |
|---|---|---|---|
| Induction | Western blot, immunofluorescence | p-ULK1 (Ser757), p-mTOR | Increased p-ULK1 indicates suppressed initiation |
| Autophagosome Formation | LC3-I to LC3-II conversion, puncta formation | LC3-II levels, LC3 puncta | Increased LC3-II may indicate induction or blockage |
| Cargo Loading | Co-localization studies | p62/ubiquitin/LC3 co-localization | Increased co-localization suggests impaired degradation |
| Lysosomal Function Lysotracker staining, cathepsin activity assays | LAMP1/2, cathepsin activity, lysosomal pH | Reduced activity indicates lysosomal dysfunction | |
| Flux Completion | p62 degradation, LC3-II turnover with bafilomycin A1 | p62 accumulation, LC-II turnover ratio | p62 accumulation with blocked lysosomal degradation indicates upstream defects |
Experimental Assessment of Autophagic Flux
Proper assessment of autophagic flux requires multiple complementary approaches, as static measurements of autophagy markers can be misleading. The following protocol provides a comprehensive framework for evaluating autophagic flux:
Protocol 2: Comprehensive Autophagic Flux Assessment
LC3 Turnover Assay:
- Treat cells with lysosomal inhibitors (100 nM bafilomycin A1 or 20 mM ammonium chloride) for 4-6 hours
- Prepare cell lysates and perform western blot for LC3
- Calculate flux as: LC3-II (with inhibitor) - LC3-II (without inhibitor)
- Higher differences indicate active flux; minimal differences suggest pre-existing blockage
p62 Degradation Assay:
- Monitor p62/SQSTM1 levels by western blot under basal conditions and after autophagy induction (e.g., serum starvation)
- Accumulation of p62 under basal conditions suggests impaired autophagic degradation
- Note: p62 is primarily degraded by autophagy, as prolonged autophagy inhibition (96h) significantly increases p62 in cortical neurons [75]
TFEB Localization Studies:
- Fix cells and perform immunofluorescence for TFEB
- Score nuclear vs. cytoplasmic localization (>70% nuclear localization indicates activation)
- Correlate with lysosomal gene expression (cathepsin D, LAMP1) via qRT-PCR
Long-lived Protein Degradation Assay:
- Label proteins with [³H]-leucine for 24-48 hours
- Chase with excess unlabeled leucine for 4-24 hours
- Measure acid-soluble radioactivity in medium as indicator of protein degradation
- Inhibit autophagy with 10 mM 3-methyladenine to determine autophagy-specific contribution
Comparative Analysis of Failure Modes
The failure mechanisms of UPS and autophagy, while distinct, exhibit significant interconnectivity in protein aggregation diseases. The following diagrams illustrate the key failure points in each system and their interrelationships:
Figure 1: Interplay between UPS inhibition and autophagic blockage in protein aggregation. Protein aggregates directly inhibit UPS function, leading to compensatory autophagy induction, which may become overwhelmed, resulting in complete degradation failure and further aggregate accumulation.
Figure 2: Specific failure mechanisms in UPS and autophagy. Protein aggregates impact both systems through distinct but complementary mechanisms, with UPS failure primarily involving physical and competitive inhibition, while autophagic failure involves breakdowns in the multi-step flux process.
Table 3: Comparative Analysis of UPS vs. Autophagy Failure Characteristics
| Characteristic | UPS Failure | Autophagic Flux Blockage |
|---|---|---|
| Primary substrates affected | Short-lived soluble proteins, regulatory proteins | Long-lived proteins, protein aggregates, damaged organelles |
| Key failure triggers | Protein aggregates, proteasome inhibitors, oxidative stress | Lysosomal dysfunction, adaptor protein mutations, fusion defects |
| Temporal progression | Often rapid inhibition by aggregates | Typically slower, progressive dysfunction |
| Compensatory response | Upregulation of autophagy via TFEB activation | Limited UPS upregulation capacity |
| Reversibility potential | Potentially reversible if aggregates are cleared | Often progressive without external intervention |
| Experimental assessment | Ubiquitinated protein accumulation, proteasome activity assays | LC3/p62 turnover, lysosomal function tests |
| Therapeutic implications | Aggregation inhibitors, proteasome activators | Autophagy enhancers, lysosomal agents |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Studying UPS and Autophagy Failures
| Reagent Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| UPS Inhibitors | Epoxomicin (1-10 μM), MG132 (5-20 μM) | Selective proteasome inhibition | Positive controls for UPS impairment; monitor ubiquitinated proteins |
| Autophagy Inhibitors | Bafilomycin A1 (50-100 nM), Chloroquine (20-100 μM) | Lysosomal acidification blockade | Flux measurements; can secondarily affect proteasomes at high doses |
| Autophagy Inducers | Rapamycin (0.1-1 μM), Torin1 (250 nM) | mTOR inhibition, autophagy activation | Test compensatory capacity; assess flux under stimulated conditions |
| Activity Reporters | LLVY-AMC, LLR-AMC, LLE-AMC | Proteasome activity substrates | Fluorogenic measurements of different proteasome activities |
| Key Antibodies | Anti-ubiquitin (FK2), anti-LC3, anti-p62, anti-TFEB | Detection of pathway components | Essential for western blot, immunofluorescence; validate specificity |
| LysoTracker Dyes | LysoTracker Red DND-99, LysoSensor Yellow/Blue | Lysosomal pH and mass indicators | Live-cell imaging of lysosomal function and number |
| TFEB Translocation Reporters | TFEB-GFP constructs, immunostaining | Monitor TFEB nuclear localization | Indicator of lysosomal stress and compensatory activation |
The failure points of the ubiquitin-proteasome system and autophagy represent critical junctures in the progression of protein aggregation diseases. UPS vulnerability stems primarily from its architectural limitations against aggregated proteins and the potential for direct inhibition, while autophagic failure typically involves breakdowns in the multi-stage flux process. The interconnectedness of these systems creates vulnerability to vicious cycles where impairment in one system overloads the other, leading to complete proteostatic collapse. Understanding these specific failure mechanisms provides not only insight into disease pathogenesis but also reveals strategic opportunities for therapeutic intervention. Future research directions should focus on developing quantitative metrics for failure progression, identifying early biomarkers of incipient dysfunction, and creating targeted approaches to reinforce each system's vulnerable points against the escalating challenge of protein aggregation.
The ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) represent the two major intracellular protein degradation systems essential for cellular homeostasis. The UPS primarily degrades short-lived, soluble proteins, while autophagy handles larger substrates like protein aggregates and damaged organelles [1]. Historically viewed as independent, emerging research reveals intricate compensatory crosstalk between these pathways, particularly when one system is impaired. This reciprocal relationship, where proteasome inhibition activates autophagy and vice versa, represents a critical adaptive mechanism with profound implications for diseases ranging from cancer to neurodegenerative disorders [38] [78] [77]. Understanding this dynamic interplay provides crucial insights for therapeutic interventions targeting proteostasis.
Molecular Mechanisms of UPS-to-Autophagy Signaling
TFEB: A Master Regulator of Compensatory Crosstalk
When proteasome activity is compromised, cells rapidly activate autophagy through Transcription Factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagic genes. Under normal conditions, phosphorylated TFEB remains sequestered in the cytoplasm. Proteasome inhibition promotes TFEB dephosphorylation and nuclear translocation, where it upregulates genes involved in autophagosome formation and lysosomal function [77].
Mechanistic Insights: Research demonstrates that TFEB itself is degraded via the proteasome pathway, with a half-life of approximately 13.5 hours in neuronal-like cells. Proteasome impairment not only stabilizes TFEB but also activates it through dephosphorylation, creating a powerful feedback loop that enhances autophagic capacity [77]. This TFEB-mediated response to proteasome inhibition significantly increases expression of key autophagic markers including LC3-II, cathepsin D, and LAMP1 [77].
Ubiquitin Signaling as a Molecular Bridge
The ubiquitin code serves as a critical language coordinating degradation decisions between the UPS and autophagy. The nature of polyubiquitin chains often determines the degradation route:
- K48-linked ubiquitin chains: Typically target substrates for proteasomal degradation [1]
- K63-linked ubiquitin chains: Often signal for autophagic clearance [1]
Proteasome inhibition causes accumulation of K48-linked ubiquitinated proteins, which can be remodeled to K63 linkages, redirecting substrates to autophagy [79]. Adaptor proteins like p62/SQSTM1 contain both ubiquitin-associated (UBA) domains and LC3-interacting regions (LIR), physically bridging ubiquitinated cargo to the autophagic machinery [1].
Proteaphagy: Autophagic Clearance of Proteasomes
Under sustained proteasome inhibition, cells can activate proteaphagy - the selective autophagic degradation of proteasomes themselves. This process, mediated by receptors like p62, represents an extreme form of crosstalk where autophagy regulates the abundance of proteasomal complexes [79]. In bortezomib-resistant mantle cell lymphoma, constitutively activated proteaphagy contributes to drug resistance by reducing intracellular proteasome levels [79].
Table 1: Key Molecular Mediators of UPS-to-Autophagy Crosstalk
| Mediator | Function | Effect of Proteasome Inhibition |
|---|---|---|
| TFEB | Master transcriptional regulator of autophagy and lysosomal genes | Dephosphorylation, nuclear translocation, and increased expression |
| p62/SQSTM1 | Autophagy receptor linking ubiquitinated cargo to LC3 | Increased accumulation and enhanced substrate recruitment |
| K63 Ubiquitin Chains | Alternative ubiquitin linkage signaling autophagy | Increased formation and substrate redirection |
| TRIM24 | E3 ubiquitin ligase regulating UPS/autophagy balance | Altered stability and subcellular localization |
Experimental Evidence: Documenting the Crosstalk
Pharmacological Inhibition Studies
Studies using specific proteasome inhibitors provide compelling evidence for UPS-autophagy crosstalk:
MG132 and Bortezomib Models: Treatment with these proteasome inhibitors consistently activates autophagic flux across multiple cell types. In neuronal-like cells, MG132 treatment increases LC3-II conversion and promotes autophagosome biogenesis without impairing autophagic flux [77]. The combination of MG132 with propolin G synergistically induces proteotoxic stress and apoptosis in breast cancer cells, with autophagy activation serving as a key cell death mechanism [80].
Experimental Protocol - Proteasome Inhibition-Induced Autophagy:
- Cell Treatment: Apply proteasome inhibitor (e.g., 1μM MG132 or 10-20nM bortezomib) for 4-24 hours
- Autophagy Assessment: Monitor LC3-I to LC3-II conversion via western blot
- Lysosomal Activity: Measure cathepsin D expression and LAMP1/2 levels
- TFEB Localization: Track nuclear translocation via immunofluorescence
- Functional Assays: Assess autophagic flux using LC3 turnover assays with lysosomal inhibitors (bafilomycin A1)
Cancer Resistance Models
Research in bortezomib-resistant mantle cell lymphoma (MCL) reveals sophisticated adaptive mechanisms. Resistant cells exhibit constitutive proteaphagy mediated by p62, effectively reducing intracellular proteasome concentration [79]. The E3 ubiquitin ligase TRIM24 emerges as a pivotal regulator in this process, influencing the balance between K48 and K63 ubiquitin chain formation. Genetic knockout of TRIM24 or pharmacological reduction using PROTAC technology restores bortezomib sensitivity, demonstrating the therapeutic potential of manipulating this crosstalk [79].
Table 2: Experimental Models of UPS-Autophagy Crosstalk
| Experimental System | Intervention | Observed Autophagy Activation | Key Findings |
|---|---|---|---|
| Neuronal-like cells | MG132 (1-10μM) | Increased LC3-II, lysosomal biogenesis | TFEB dephosphorylation and nuclear translocation |
| Breast cancer cells | MG132 + Propolin G | Synergistic activation (CI=0.63) | PERK/ATF4/CHOP UPR pathway activation |
| Mantle cell lymphoma | Bortezomib (acquired resistance) | Constitutive proteaphagy | TRIM24-dependent K63 ubiquitin chain enrichment |
| Arabidopsis model | MG132 treatment | ATG8a stabilization via N-degron pathway | Arg/N-degron pathway governs ATG8a turnover |
Autophagy-to-UPS Regulation: The Reverse Signal
Autophagy Regulation of Proteasome Homeostasis
The crosstalk between degradation pathways is bidirectional. Autophagy influences UPS function through several mechanisms:
Proteaphagy: As mentioned, autophagy selectively degrades proteasomes under certain conditions, directly regulating UPS capacity [79]. In Arabidopsis, this process is mediated by regulatory particle non-ATPase 10 (RPN10), which recognizes inactive proteasomes for autophagic clearance [1].
Transcription Factor Crosstalk: Beyond TFEB, other transcription factors like FOXO and E2F coordinate the expression of both proteasomal and autophagic components, creating coordinated transcriptional programs that balance the two systems [77].
The N-degron Pathway Connection
Recent research in Arabidopsis reveals the Arg/N-degron pathway as a crucial regulator of autophagy components, directly linking ubiquitin-mediated degradation to autophagic function. The core autophagy component ATG8a is targeted for proteasomal degradation via the Arg/N-degron pathway, with ubiquitin ligase E3 component N-recognin 7 (UBR7) responsible for its ubiquitination and degradation [42]. This pathway creates a feedback loop where autophagy regulates the degradation of its own components through the UPS.
Research Reagent Solutions
Table 3: Essential Research Tools for Studying UPS-Autophagy Crosstalk
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib (BTZ) | Induce proteotoxic stress | Inhibit chymotrypsin-like activity of 20S proteasome |
| Autophagy Inhibitors | Bafilomycin A1 (BafA1), Chloroquine | Block autophagic flux | Prevent lysosomal acidification and degradation |
| Lysosomal Inhibitors | Concanamycin A | Distinguish degradation pathways | Specific vacuolar H+-ATPase inhibitor |
| Autophagy Inducers | Rapamycin, Torin1 | Activate autophagy initiation | mTOR pathway inhibition |
| Ubiquitin Chain Probes | K48- and K63-linkage specific antibodies | Monitor ubiquitin code dynamics | Distinguish substrate targeting signals |
| TFEB Translocation Reporters | TFEB-GFP constructs, anti-TFEB antibodies | Track master regulator activation | Monitor subcellular localization |
| Autophagy Receptors | p62/SQSTM1, NBR1 constructs | Study selective autophagy | Bridge ubiquitinated cargo to LC3 |
| PROTAC Degraders | dTRIM24 | Target specific E3 ligases | Pharmacologically reduce TRIM24 levels |
The reciprocal regulation between the UPS and autophagy represents a fundamental homeostatic mechanism with significant therapeutic implications. In cancer, manipulating this crosstalk may overcome resistance to proteasome inhibitors [79]. In neurodegenerative diseases, enhancing the compensatory activation of autophagy may help clear toxic protein aggregates [15]. Future research should focus on developing dual modulators that optimally balance these two degradation systems, potentially leveraging natural compounds like polyphenol metabolites that show UPS-modulating activity [81]. Understanding the precise molecular switches that determine substrate routing between these pathways will be crucial for designing targeted therapeutic interventions that restore proteostasis in disease states.
The ubiquitin-proteasome system (UPS) and autophagy are the two principal pillars of cellular proteostasis, responsible for the controlled degradation of proteins and organelles. The UPS primarily handles short-lived soluble proteins through a targeted, ubiquitin-dependent mechanism, while the autophagy-lysosome pathway (ALP) specializes in degrading long-lived proteins, protein aggregates, and damaged organelles such as mitochondria and endoplasmic reticulum fragments [82] [83]. In eukaryotic cells, maintaining the precise balance between these systems is crucial for cellular integrity, and their dysregulation represents a common pathological hallmark in numerous diseases, particularly neurodegenerative disorders and cancer [84] [85]. This review provides a comprehensive comparison of UPS and autophagy impairment across these disease contexts, examining their distinct yet interconnected roles in disease pathogenesis, experimental approaches for their investigation, and emerging therapeutic strategies that target these degradation pathways.
Molecular Mechanisms: UPS and Autophagy Pathways
The Ubiquitin-Proteasome System (UPS)
The UPS constitutes a highly specialized mechanism for targeted protein degradation, functioning as the primary cellular system for eliminating misfolded, damaged, or short-lived regulatory proteins. This pathway involves a coordinated enzymatic cascade: E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases work sequentially to tag substrate proteins with polyubiquitin chains [81]. These ubiquitinated substrates are then recognized and degraded by the 26S proteasome, a multi-subunit complex comprising the 20S catalytic core particle and the 19S regulatory particle that facilitates substrate deubiquitination, unfolding, and translocation into the proteolytic chamber [83]. The UPS demonstrates remarkable specificity in its degradation capacity, primarily targeting individual proteins for processing, which enables rapid regulation of protein levels and the removal of abnormal proteins before they can accumulate and cause cellular damage [55].
The Autophagy-Lysosome Pathway (ALP)
Autophagy encompasses several distinct but related pathways for bulk cytoplasmic degradation, with macroautophagy (hereafter referred to as autophagy) being the most extensively characterized in disease contexts. Autophagy proceeds through a series of orchestrated steps: initiation involving the ULK1 complex and VPS34 complex; phagophore expansion and autophagosome formation regulated by two ubiquitin-like conjugation systems (ATG12-ATG5-ATG16L and LC3-PE); and ultimately fusion with lysosomes to form autolysosomes where cargo degradation occurs [86]. This pathway demonstrates unique capacity for degrading diverse substrates, including protein aggregates, damaged organelles, and intracellular pathogens [55]. Selective forms of autophagy target specific cargoes through receptor proteins such as p62/SQSTM1, which simultaneously binds ubiquitin and LC3 to bridge ubiquitinated substrates with the growing autophagosomal membrane [55]. The autophagy process concludes with the degradation of engulfed contents by lysosomal hydrolases and recycling of resulting macromolecules back to the cytoplasm for reuse [86].
Cross-Talk and Switching Mechanisms
UPS and autophagy do not function in isolation but rather exhibit extensive cross-talk and can compensate for each other during proteotoxic stress. Research has revealed a molecular switching mechanism regulated by the BAG1/BAG3 ratio and HDAC6 levels [83]. Under normal conditions, BAG1 coordinates with HSP70 to direct substrates to the proteasome. However, during severe proteotoxic stress, such as cerebral ischemia, BAG3 expression increases while BAG1 decreases, promoting a shift toward autophagy-mediated degradation through interactions with HSP70 and HDAC6, which facilitates the transport of ubiquitinated protein aggregates along microtubules to aggresomes for autophagic clearance [83]. This switch represents a crucial adaptive response when the proteasome becomes overwhelmed, enabling cells to utilize the bulk degradation capacity of autophagy to manage accumulated protein damage.
Table 1: Comparative Features of UPS and Autophagy
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome Pathway (ALP) |
|---|---|---|
| Primary Substrates | Short-lived soluble proteins, regulatory proteins [83] | Long-lived proteins, protein aggregates, damaged organelles [82] [83] |
| Degradation Capacity | Individual proteins [55] | Bulk cytoplasm, protein aggregates, organelles [55] |
| Key Molecular Components | E1-E2-E3 enzymes, 26S proteasome [81] [83] | ULK1 complex, LC3/ATG8, p62/SQSTM1, lysosomal hydrolases [55] [86] |
| Degradation Kinetics | Rapid (minutes) | Slower (hours) |
| Energy Dependence | ATP-dependent (ubiquitination, proteasomal unfolding) [83] | ATP-dependent (autophagosome formation, fusion) |
| Selectivity Mechanism | Ubiquitin tags, E3 ligase specificity [81] | Autophagy receptors (p62, NBR1, etc.) [55] |
| Primary Regulatory Mechanisms | Ubiquitin conjugation/deconjugation, proteasome assembly [81] | mTORC1, AMPK, ATG gene expression, LC3 lipidation [86] |
Dysregulation in Neurodegenerative Diseases
Protein Aggregation and Clearance Defects
In neurodegenerative disorders, the accumulation of misfolded proteins represents a fundamental pathological feature directly linked to impairments in both UPS and autophagy function. Amyotrophic lateral sclerosis (ALS) characteristically exhibits cytoplasmic aggregates containing TAR DNA-binding protein 43 (TDP-43), whose clearance depends on functional proteasomal and autophagic systems [87] [85]. Similarly, Alzheimer's disease features amyloid-β plaques and neurofibrillary tangles, while Parkinson's disease shows α-synuclein-positive Lewy bodies, all reflecting failure of protein quality control mechanisms [85]. Quantitative expression analyses reveal that proteasome gene expression is consistently lower in the human central nervous system compared to other tissues, creating a potentially vulnerable environment for protein aggregate accumulation [85]. This inherent limitation in proteasomal capacity may predispose neuronal cells to rely more heavily on autophagy for maintaining proteostasis, particularly for handling the protein aggregates that characterize neurodegenerative pathology.
Autophagic-Lysosomal Dysfunction in Neurodegeneration
The autophagy-lysosome pathway demonstrates particular importance in neuronal health due to the post-mitotic nature of neurons and their limited capacity for protein dilution through cell division. In Alzheimer's disease, dysregulation of lysosomal autophagy is closely associated with abnormal amyloid β protein accumulation, exacerbating neuronal damage and cell death [82]. Multiple neurodegenerative conditions share common disturbances in autophagic flux, including impaired autophagosome formation, disrupted axonal transport of autophagosomes, and reduced lysosomal acidification and degradative function [55]. Lysosomal membrane stability and enzymatic activity are crucial for maintaining autophagic function, and damage to lysosomal membranes can lead to leakage of cathepsins and other hydrolases, triggering inflammatory responses that further exacerbate neuronal damage [82]. The vulnerability of autophagy-lysosomal function in aging and neurodegeneration underscores its central role in neuronal proteostasis and explains why its impairment features prominently across multiple neurodegenerative conditions.
Compensatory Mechanisms and Pathway Interdependence
The interdependence between UPS and autophagy becomes particularly evident when one system is compromised, as cells attempt to activate compensatory pathways to maintain proteostasis. Research demonstrates that when UPS function becomes impaired, either through proteasome inhibition or substrate overload, cells can activate a switch to autophagy to remove accumulating ubiquitinated proteins [83]. This adaptive response involves increased expression of BAG3, which recruits HDAC6 to facilitate the retrograde transport of protein aggregates along microtubules for autophagic degradation. However, in chronic neurodegenerative conditions, both systems often become progressively impaired, leading to loss of this compensatory capacity and accelerated disease progression. The failure of both primary degradation systems creates a vicious cycle of protein aggregation, cellular dysfunction, and ultimately neuronal death.
Table 2: UPS and Autophagy Defects in Major Neurodegenerative Diseases
| Disease | UPS Dysregulation | Autophagy Dysregulation | Key Aggregated Proteins |
|---|---|---|---|
| Alzheimer's Disease | Reduced proteasome activity, impaired ubiquitination [85] | Lysosomal dysfunction, impaired autophagosome maturation [82] | Amyloid-β, Tau [85] |
| Parkinson's Disease | Mutations in E3 ubiquitin ligase (Parkin) [82] | Impaired mitophagy, lysosomal deficiency [82] | α-Synuclein [85] |
| Amyotrophic Lateral Sclerosis | TDP-43 proteasome resistance, C9orf72-related UPS impairment [87] [85] | Impaired autophagosome formation, defective cargo recognition [87] | TDP-43, SOD1 [87] [85] |
| Huntington's Disease | Proteasome inhibition by huntingtin aggregates | Reduced autophagic activity, impaired cargo loading | Mutant huntingtin |
Dysregulation in Cancer
Proteostasis Adaptation in Malignant Cells
Cancer cells exploit both UPS and autophagy to support their survival, proliferation, and metastasis under the stressful tumor microenvironment. The UPS plays a particularly important role in degrading tumor suppressor proteins, with many cancers exhibiting overexpression of specific E3 ubiquitin ligases that target proteins like p53 for proteasomal destruction [88]. Simultaneously, cancer cells frequently upregulate autophagy to mitigate various cellular stresses, including nutrient deprivation, hypoxia, and metabolic derangements that characterize the tumor microenvironment [84]. The unfolded protein response (UPR), activated in response to endoplasmic reticulum stress, represents another proteostatic pathway co-opted by cancer cells, with IRE1α-XBP1 signaling promoting survival in aggressive malignancies such as castration-resistant prostate cancer by enhancing ER-associated degradation (ERAD) and managing the increased protein folding demands of rapid proliferation [87] [88]. This strategic manipulation of proteostasis networks enables cancer cells to thrive under conditions that would normally trigger apoptosis in non-transformed cells.
Context-Dependent Roles of Autophagy in Tumorigenesis
Autophagy demonstrates context-dependent roles in cancer, functioning as both a tumor suppression mechanism in early carcinogenesis and a pro-survival pathway in established tumors. In normal cells and early tumor development, autophagy acts as a tumor suppressor by eliminating damaged organelles, reducing oxidative stress, and preventing genomic instability [84]. However, in established tumors, autophagy promotes cancer cell survival by recycling nutrients during metabolic stress and mitigating the proteotoxic stress associated with rapid proliferation and protein synthesis [84] [88]. The dynamic regulation of autophagy throughout cancer progression presents both challenges and opportunities for therapeutic intervention, as timing, tumor type, and genetic context all influence the functional consequences of autophagy modulation. This dual nature of autophagy in cancer underscores the complexity of targeting proteostasis pathways for anticancer therapy.
UPS and Autophagy as Therapeutic Targets in Oncology
Both UPS and autophagy represent promising therapeutic targets in cancer, with multiple intervention strategies currently under investigation. Proteasome inhibitors such as bortezomib, carfilzomib, and ixazomib have achieved clinical success in hematological malignancies, particularly multiple myeloma, by inducing ER stress and apoptosis through the accumulation of misfolded proteins [88]. Autophagy inhibition with hydroxychloroquine or related compounds is being explored in clinical trials to sensitize tumor cells to conventional chemotherapy and targeted therapies, particularly in tumors with high basal autophagy [84]. Additionally, novel approaches targeting the UPR pathway are under development, including IRE1α RNase inhibitors (STF-083010, 4μ8C) and PERK inhibitors (GSK2606414), which have shown efficacy in preclinical cancer models [87]. The emerging understanding of proteostasis networks in cancer continues to reveal new vulnerabilities that can be exploited therapeutically.
Experimental Approaches and Methodologies
Assessing UPS and Autophagy Activity
Investigating UPS and autophagy function requires specific methodological approaches that can accurately measure the activity and flux through these pathways. For monitoring UPS function, researchers commonly employ reporter substrates such as ubiquitin-GFP, measure proteasome peptidase activities using fluorogenic substrates (e.g., Suc-LLVY-AMC), and quantify levels of polyubiquitinated proteins through western blotting [83]. Autophagy assessment typically involves measuring the conversion of LC3-I to LC3-II by immunoblotting, quantifying autophagic flux using tandem fluorescent LC3 constructs (such as mRFP-GFP-LC3) that differentiate between autophagosomes and autolysosomes based on pH sensitivity, and monitoring p62/SQSTM1 degradation as an indicator of functional autophagic completion [55] [86]. These complementary approaches allow researchers to distinguish between mere accumulation of autophagic structures versus genuine increases in autophagic degradation activity, a critical distinction for accurately interpreting experimental results in both physiological and pathological contexts.
Modeling Protein Aggregation and Clearance
Experimental models of protein aggregation and clearance employ various strategies to induce and monitor proteostasis disruption. Cerebral ischemia models using transient middle cerebral artery occlusion (tMCAO) in mice have demonstrated accelerated ubiquitin-positive protein aggregation during reperfusion, with a subsequent switch from UPS to autophagy that depends on ischemic duration and involves increased expression of BAG3 and HDAC6 alongside decreased BAG1 [83]. Cellular models often utilize fluorescently tagged aggregation-prone proteins (e.g., mutant huntingtin, TDP-43, or α-synuclein) to monitor aggregate formation and clearance in real time [55]. These experimental systems enable detailed investigation of the molecular mechanisms underlying UPS-autophagy cross-talk and provide platforms for screening potential therapeutic compounds that enhance clearance of pathological protein aggregates.
Table 3: Key Experimental Approaches for Studying UPS and Autophagy
| Method Category | Specific Techniques | Key Readouts | Considerations |
|---|---|---|---|
| UPS Activity Assays | Fluorogenic proteasome substrates, Ubiquitin-protein pulldowns, Proteasome activity gels [83] | Proteasome catalytic activity, Ubiquitinated protein levels, Proteasome composition | Distinguish 26S vs 20S proteasome activity; consider ATP-dependence |
| Autophagy Flux Measurements | LC3-I/LC3-II immunoblotting, Tandem fluorescent LC3 reporters, p62 degradation assays [55] [86] | LC3-II:LC3-I ratio, Autophagosomes vs autolysosomes, p62 clearance | Use multiple methods; always measure flux, not just markers |
| Pathway Switching Studies | BAG1/BAG3 ratio measurement, HDAC6 inhibition, Ubiquitin aggregate tracking [83] | BAG1/BAG3 protein levels, Aggregate clearance kinetics, Colocalization studies | Consider stress duration and intensity; multiple timepoints |
| Genetic Manipulations | siRNA/shRNA knockdown, CRISPR/Cas9 knockout, Dominant-negative constructs | Pathway component expression, Compensatory pathway activation | Account for potential compensatory mechanisms |
Research Reagent Solutions
Chemical Inhibitors and Activators
The study of UPS and autophagy relies heavily on specific pharmacological tools that selectively modulate these pathways. For UPS inhibition, researchers commonly use MG132, bortezomib, and epoxomicin, which directly target proteasomal catalytic activities [83]. Autophagy modulation includes inhibitors such as chloroquine and hydroxychloroquine (which prevent autophagosome-lysosome fusion), 3-methyladenine (which blocks class III PI3K and autophagosome formation), and bafilomycin A1 (which inhibits lysosomal acidification) [86]. Conversely, autophagy can be induced using rapamycin and related mTOR inhibitors, which relieve suppression of autophagy initiation, or Torin1, which more completely inhibits mTORC1 [86]. For investigating the UPR pathway, specific inhibitors are available including STF-083010 and 4μ8C (targeting IRE1α RNase activity) and GSK2606414 (targeting PERK) [87]. These pharmacological tools enable precise dissection of pathway contributions to proteostasis maintenance in both health and disease.
Molecular and Cellular Biology Tools
Advanced molecular tools have significantly enhanced our ability to study UPS and autophagy dynamics in live cells and model organisms. Fluorescent reporters including ubiquitin-GFP for monitoring protein turnover, and the mRFP-GFP-LC3 tandem construct for tracking autophagic flux through the differential pH stability of RFP versus GFP, provide real-time information on pathway activity [55]. CRISPR/Cas9 technology enables generation of knockout cell lines for specific autophagy-related genes (ATGs), ubiquitin ligases, or proteasome subunits to determine their essential roles in proteostasis [55]. Antibody-based reagents remain crucial for techniques such as immunoblotting (e.g., anti-LC3, anti-ubiquitin, anti-p62 antibodies) and immunohistochemistry to assess spatial distribution of pathway components and substrates in tissues and fixed cells [83] [86]. These research tools continue to evolve, offering increasingly sophisticated approaches for investigating the complex interplay between UPS and autophagy in pathological conditions.
Signaling Pathway Visualizations
Unfolded Protein Response Signaling Pathway
UPR Signaling Pathway Activation: This diagram illustrates the three primary branches of the unfolded protein response (UPR) activated by endoplasmic reticulum (ER) stress. The pathway begins with the accumulation of misfolded proteins in the ER, leading to BiP/GRP78 release and subsequent activation of PERK, IRE1α, and ATF6 sensors. Each arm orchestrates distinct transcriptional and translational responses that collectively determine cellular fate, enabling either adaptation and restored homeostasis or apoptosis under irremediable ER stress conditions [87] [88].
UPS to Autophagy Switching Mechanism
UPS to Autophagy Switching Mechanism: This diagram depicts the molecular switch from ubiquitin-proteasome system (UPS) to autophagy during proteotoxic stress. Under mild stress, BAG1 coordinates with HSP70 to direct substrates to the proteasome. However, when UPS becomes overloaded during severe stress, BAG3 expression increases while BAG1 decreases, promoting recruitment of HDAC6 and facilitating aggregate transport to aggresomes for autophagic clearance [83].
Concluding Perspectives
The comparative analysis of UPS and autophagy impairment in neurodegeneration and cancer reveals both shared and distinct pathophysiological mechanisms across these disease contexts. In neurodegenerative disorders, both systems typically become progressively impaired, leading to accumulation of toxic protein aggregates and neuronal dysfunction. In contrast, cancer cells often exploit these pathways to support survival and growth under stress, though the specific adaptations vary considerably between cancer types and stages. The emerging understanding of the molecular mechanisms governing the interplay and switching between UPS and autophagy provides novel insights for therapeutic development. Targeted protein degradation technologies, including PROTACs that harness the UPS and emerging autophagy-targeting approaches such as AUTAC, ATTEC, and AUTOTAC, represent promising strategies for selectively eliminating disease-causing proteins in both neurological disorders and cancer [55]. Future research directions should focus on elucidating the precise molecular determinants of pathway selection, developing more sophisticated methods for monitoring UPS and autophagy activity in human patients, and designing context-specific therapeutic interventions that can selectively modulate these pathways to restore proteostasis in disease states.
The degradation of misfolded proteins is a critical cellular process governed primarily by two major systems: the ubiquitin-proteasome system (UPS) and autophagy. The UPS is highly specific and degrades soluble, short-lived proteins through a sophisticated enzymatic cascade involving ubiquitination and proteasomal cleavage [10] [89]. In contrast, autophagy, particularly macroautophagy and chaperone-mediated autophagy (CMA), handles larger substrates, including protein aggregates and damaged organelles, through lysosomal degradation [55] [89]. For researchers and drug development professionals, understanding the interplay, strengths, and limitations of these pathways is fundamental to developing novel targeted protein degradation (TPD) technologies. This guide objectively compares key strategies that leverage these endogenous systems, providing a structured analysis of their mechanisms, experimental support, and optimization potential.
Comparative Analysis of Major Degradation Strategies
The following table summarizes the core characteristics, advantages, and limitations of major technologies that leverage the UPS and autophagy pathways.
Table 1: Comparison of Key Targeted Protein Degradation Technologies
| Technology | Primary Degradation Pathway | Key Components & Mechanism | Therapeutic Advantages | Major Limitations |
|---|---|---|---|---|
| PROTACs [43] [55] | Ubiquitin-Proteasome System (UPS) | Heterobifunctional molecule with a target protein ligand, an E3 ubiquitin ligase ligand, and a linker. Recruits E3 ligase to ubiquitinate the target protein, leading to proteasomal degradation. | High selectivity and potency; catalytic mode of action; can target undruggable proteins (e.g., transcription factors). | Poor solubility and bioavailability; limited to intracellular proteins; potential for resistance due to proteasome insufficiency [55]. |
| Molecular Glues [43] | Ubiquitin-Proteasome System (UPS) | Monovalent small molecules that induce or stabilize the interaction between an E3 ligase and a target protein, leading to ubiquitination and degradation. | Often have superior drug-like properties compared to bifunctional degraders; can engage targets without a known ligand. | Discovery is largely serendipitous; rational design is challenging. |
| Trim-Away [90] | Ubiquitin-Proteasome System (UPS) | Utilizes antibodies against the native target protein and the E3 ubiquitin ligase TRIM21. The antibody binds the target, TRIM21 binds the antibody's Fc region, and ubiquitinates the complex for proteasomal degradation. | Degrades unmodified, endogenous proteins rapidly (within minutes); applicable to a wide range of proteins and cell types, including primary non-dividing cells. | Requires delivery of antibody and often TRIM21; efficiency can be cell-type dependent; potential immunogenicity. |
| AUTAC [43] [55] | Autophagy-Lysosome Pathway (Macroautophagy) | Bifunctional degrader with a target-binding ligand linked to a degradation tag (e.g., guanine derivative) that mimics an S-guanylation signal, targeting the protein to autophagosomes. | Can degrade large protein aggregates and organelles; useful for targets resistant to UPS degradation. | Degradation is relatively slower than UPS-based methods; the specific E3 ligases and mechanisms for tagging are still under investigation. |
| ATTEC [55] | Autophagy-Lysosome Pathway (Macroautophagy) | Small molecules that simultaneously bind to the target protein (e.g., mutant huntingtin) and the autophagosome-associated protein LC3, tethering the target directly to the autophagosome for degradation. | Direct tethering to the core autophagy machinery; potential high specificity and efficiency for specific targets. | Requires discovery of dual-binding molecules for each target; limited to targets with known ATTEC compounds. |
| CMA-Targeting Peptides [91] | Chaperone-Mediated Autophagy (CMA) | Cell-penetrant peptide with a Protein Binding Domain (PBD), a CMPD for membrane penetration, and a CMA-Targeting Motif (CTM, e.g., KFERQ-like sequence). The peptide binds the target and shuttles it to lysosomes via the LAMP2A receptor. | Rapid and reversible knockdown; high specificity for the target protein; works in vivo (e.g., can cross blood-brain barrier). | Requires a specific PBD for each target; efficiency depends on endogenous CMA activity, which declines with age. |
Core Mechanisms and Signaling Pathways
The efficacy of degradation technologies hinges on their engagement with specific components of the UPS or autophagy pathways. The following diagrams illustrate the fundamental mechanisms of these two systems and how key technologies interface with them.
Ubiquitin-Proteasome System (UPS) and Key Technologies
The UPS is the primary pathway for degrading soluble, misfolded, and short-lived regulatory proteins. Degradation involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that tag the target protein with a polyubiquitin chain, primarily via K48 linkage. This chain signals the 26S proteasome to unfold, deubiquitinate, and proteolyze the target into short peptides [10] [89]. Technologies like PROTACs and Trim-Away are designed to hijack this specific and rapid pathway.
Diagram Title: UPS Mechanism and Degrader Action
Autophagy-Lysosome Pathway and Key Technologies
Autophagy is a bulk degradation pathway capable of clearing large protein aggregates and organelles. Key types include macroautophagy, where a double-membraned autophagosome engulfs cargo and fuses with a lysosome, and chaperone-mediated autophagy (CMA), where proteins with a KFERQ motif are directly translocated across the lysosomal membrane via the LAMP2A receptor [55] [89]. Technologies like AUTAC, ATTEC, and CMA-targeting peptides are engineered to leverage these mechanisms with varying degrees of selectivity.
Diagram Title: Autophagy Pathways and Degrader Action
Experimental Protocols and Supporting Data
Protocol 1: Acute Protein Degradation Using Trim-Away
Trim-Away is a powerful technique for the rapid degradation of endogenous, unmodified proteins, ideal for functional studies in primary and non-dividing cells [90].
- Key Reagents: (1) Antibody: High-affinity antibody against the native target protein. (2) TRIM21: Purified recombinant TRIM21 protein or expression vector. (3) Delivery System: Microinjection equipment or electroporation for co-delivery of antibody and TRIM21 into cells.
- Methodology:
- Preparation: Dilute the target-specific antibody and TRIM21 (if using protein) in a suitable injection buffer.
- Delivery: Co-microinject the antibody and TRIM21 into the cytoplasm of target cells. Controls should include injection of a non-specific IgG.
- Incubation & Analysis: Incubate cells at 37°C. Protein degradation can be observed within minutes to hours. Monitor degradation efficiency via western blotting or immunofluorescence, comparing to non-injected or control-injected cells.
- Key Experimental Data: In a proof-of-concept study, microinjection of an anti-GFP antibody into NIH 3T3 cells overexpressing mCherry-TRIM21 and GFP led to rapid GFP degradation with a half-life of approximately 16 minutes. Degradation was dependent on the E3 ligase activity of TRIM21 and was completely blocked by the proteasome inhibitor MG132 [90].
Protocol 2: Targeted Degradation via Autophagy (AUTAB)
The recently developed AUTophagy-inducing Antibody (AUTAB) technology targets extracellular and membrane proteins for degradation by the autophagy-lysosome pathway [55].
- Key Reagents: (1) AUTAB Antibody: A monoclonal antibody against a cell surface target, engineered to be an agonist of the IGF1 receptor (IGF1R). (2) Cell Culture: Target cells expressing the protein of interest and IGF1R.
- Methodology:
- Treatment: Incubate cells with the AUTAB antibody. A control should include an isotype control antibody.
- Activation: AUTAB binding to IGF1R induces its autophosphorylation and internalization, triggering downstream signaling.
- Autophagy Induction: The activated IGF1R signaling cascade induces macroautophagy.
- Degradation & Analysis: The internalized target-AUTAB complex is trafficked to autolysosomes for degradation. Analyze target protein levels by western blot or flow cytometry post-treatment (e.g., 4-24 hours). Monitor autophagic flux by tracking LC3-I to LC3-II conversion and p62/SQSTM1 degradation [55].
- Key Experimental Data: Proof-of-concept studies targeting PD-L1 showed that AUTACs induced autophagic degradation of the target. The technology leverages the cell's natural receptor signaling to induce degradation, offering a novel approach for cell surface proteins [55].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their critical functions in the experiments and technologies discussed.
Table 2: Key Research Reagents for Targeted Degradation Studies
| Research Reagent | Function in Experiment/Degradation Process |
|---|---|
| E3 Ubiquitin Ligases (e.g., VHL, CRBN, TRIM21) | Central to UPS-based strategies (PROTACs, Molecular Glues, Trim-Away). They confer substrate specificity by recognizing the target and catalyzing its ubiquitination [10] [90]. |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Used to experimentally validate UPS-dependent degradation. The blockade of degradation by these inhibitors confirms the involvement of the proteasome [90] [89]. |
| Lysosome Inhibitors (e.g., Ammonium Chloride, Chloroquine, Bafilomycin A1) | Used to distinguish lysosomal degradation from proteasomal degradation. They neutralize lysosomal pH, inhibiting hydrolase activity and blocking both macroautophagy and CMA [91]. |
| LC3 (Microtubule-associated protein 1A/1B-light chain 3) | A key marker for autophagosomes. The conversion from cytosolic LC3-I to lipidated, autophagosome-bound LC3-II (detected by western blot) and its punctate formation (seen by immunofluorescence) are gold standards for monitoring autophagy [55]. |
| p62/SQSTM1 (Sequestosome-1) | A selective autophagy receptor that links ubiquitinated proteins to LC3. The level of p62 decreases when autophagic flux is complete; its accumulation indicates blocked autophagy [55] [89]. |
| LAMP2A (Lysosomal-associated membrane protein 2A) | The critical receptor for CMA. Its levels at the lysosomal membrane are rate-limiting for CMA activity. Used to assess CMA capability in cells [91] [89]. |
| CMA-Targeting Motif (CTM) Peptides | Synthetic peptides containing a KFERQ-like sequence. Used to hijack the CMA pathway to shuttle bound target proteins to the lysosome for degradation [91]. |
| Cell-Penetrating Peptides (CPP) | Often incorporated into degrader designs (e.g., CMA-targeting peptides) to facilitate delivery across the plasma membrane and, in some cases, the blood-brain barrier [91]. |
Targeted Protein Degradation (TPD) represents a paradigm shift in therapeutic strategy, moving beyond simple inhibition to the complete elimination of disease-causing proteins. However, the journey from a potent degrader molecule to an effective drug is fraught with challenges related to poor solubility, limited bioavailability, and inefficient tissue-specific delivery. This guide provides a structured comparison of how these hurdles impact different TPD modalities and outlines the experimental frameworks used to develop solutions.
TPD technologies harness the cell's innate protein degradation machinery, primarily the Ubiquitin-Proteasome System (UPS) and the Autophagy-Lysosome Pathway, to eliminate pathological proteins [67] [68]. Proteolysis-Targeting Chimeras (PROTACs) are the most advanced UPS-engaging technology, while autophagy-targeting chimeras (AUTACs, ATTECs, AUTOTACs) represent a growing class of lysosome-directed degraders [43] [55]. Despite their therapeutic promise, these agents face significant delivery obstacles. Their often large molecular weight and high lipophilicity can lead to poor solubility and limited cellular permeability [67] [92]. Furthermore, achieving catalytic activity requires the degrader to successfully enter the cell, form a productive ternary complex, and then escape to facilitate multiple degradation cycles [92]. This review objectively compares the delivery and bioavailability profiles of major TPD strategies, providing a foundational toolkit for their advancement.
Comparative Analysis of TPD Strategies and Delivery Hurdles
The core mechanisms of UPS and autophagy-based degraders inherently influence their pharmacological profiles and the specific delivery challenges they encounter. The table below provides a high-level comparison of the two major TPD strategic classes.
Table 1: Strategic Comparison of Major TPD Pathways
| Feature | UPS-Based TPD (e.g., PROTACs) | Autophagy-Based TPD (e.g., AUTACs, ATTECs) |
|---|---|---|
| Primary Degradation Machinery | 26S Proteasome [27] [93] | Autophagy-Lysosome System [68] [55] |
| Typical Substrate Scope | Short-lived, soluble proteins [27] | Protein aggregates, organelles, long-lived proteins [68] |
| Key Delivery Challenge | Hook effect at high concentrations [67] | Achieving selectivity for specific targets within a bulk degradation process [55] |
| Molecular Size | Often large (>700 Da) [92] | Variable; can be smaller (e.g., ATTECs) [55] |
| Oral Bioavailability | Demonstrated for some clinical candidates (e.g., ARV-110, ARV-471) [93] | Largely preclinical; oral potential under investigation [68] |
A deeper analysis of specific technologies reveals critical distinctions in their properties and the experimental data surrounding their delivery.
Table 2: Experimental Degradation Profile and Delivery Characteristics of Key TPD Modalities
| TPD Modality (Example) | Reported DC₅₀ / Efficacy | Key Delivery & Bioavailability Findings | Experimental Model |
|---|---|---|---|
| PROTAC (ARV-110) | DC₅₀ ~1 nM for AR degradation [93] | Achieved tumor degradation in vivo; no dose-limiting toxicity up to 420 mg in clinical trials [93] | Prostate cancer cell lines; mCRPC patients [93] |
| PROTAC (KT-253) | >200-fold more potent than MDM2 inhibitor [67] | Sustained tumor regression in xenograft models; prompted Phase I clinical trial [67] | Xenograft models [67] |
| AUTAC | Degrades fragmented mitochondria and proteins like BRD4 [68] | Utilizes a guanine derivative tag for degradation signaling; scope includes organelles [68] | Cell-based assays [68] |
| ATTEC | Directly binds both target (e.g., mHTT) and LC3 protein on autophagosome [68] [55] | Smaller molecule size may improve permeability; demonstrated target engagement in neurodegenerative models [68] [55] | Cell-based models of Huntington's disease [68] |
Visualizing Core Degradation Pathways and Experimental Workflows
The efficacy of any TPD therapeutic is contingent upon its successful engagement of a specific degradation pathway. The following diagrams map these core mechanisms and a standard workflow for evaluating degrader efficacy.
The Ubiquitin-Proteasome System (UPS) Pathway
The Autophagy-Lysosome Pathway
Experimental Workflow for TPD Efficacy and Delivery Assessment
Essential Protocols for Evaluating TPD Delivery and Efficacy
To objectively compare TPD therapeutics, standardized experimental protocols are essential. Below are detailed methodologies for key assays cited in the literature.
Protocol: Determining Degradation Potency (DC₅₀) and Hook Effect
This protocol is fundamental for establishing the core activity and optimal dosing range of a TPD therapeutic [67] [92].
- Cell Seeding and Treatment: Seed appropriate target cells (e.g., LNCaP for AR degradation) in 96-well plates. Allow cells to adhere for 24 hours.
- Dose-Response Curve: Treat cells with the TPD degrader (e.g., a PROTAC) across a broad concentration range (e.g., 1 pM to 10 µM) for a predetermined time (typically 4-24 hours).
- Hook Effect Assessment: Ensure the highest concentration points are sufficiently high (e.g., >10 µM for many PROTACs) to observe potential self-inhibition due to the formation of non-productive binary complexes [67].
- Cell Lysis and Analysis: Lyse cells and quantify the target protein level using a validated method, such as:
- Western Blotting: Semi-quantitative analysis of protein bands.
- Cellular Thermal Shift Assay (CETSA): Confirm target engagement.
- Immunofluorescence: Visualize protein loss and subcellular localization.
- Data Calculation: Normalize protein levels to a loading control (e.g., GAPDH) and vehicle-treated cells. Plot the percentage of protein remaining versus the log of the degrader concentration. The DC₅₀ is the concentration at which 50% of the target protein is degraded.
Protocol: Assessing Ternary Complex Formation
The formation of a stable POI-degrader-E3 ligase complex is a critical event for UPS-based degraders [92]. This can be assessed via:
- Surface Plasmon Resonance (SPR): Immobilize the E3 ligase on a sensor chip. Inject a pre-formed mixture of the POI and the degrader over the surface. A stronger response signal compared to injecting the POI alone indicates stabilization of the ternary complex.
- Co-Immunoprecipitation (Co-IP): Treat cells expressing tagged versions of the POI and E3 ligase with the degrader. Lyse cells and immunoprecipitate the POI. Probe the immunoprecipitate for the presence of the E3 ligase via Western blot to confirm induced proximity [94].
- X-ray Crystallography/Cryo-EM: For definitive structural insight, the ternary complex can be purified and its structure solved, providing atomic-level data on binding interfaces [92].
Protocol: Evaluating Autophagic Flux in Autophagy-Based TPD
For degraders utilizing the autophagy pathway (e.g., AUTACs, ATTECs), confirming activity through the intended pathway is crucial [68] [55].
- LC3-I/LC3-II Conversion Assay: Treat cells with the autophagy-based degrader. Analyze cell lysates by Western blot using an anti-LC3 antibody. A increase in the lipidated form (LC3-II) relative to the cytosolic form (LC3-I) indicates autophagosome formation.
- p62/SQSTM1 Degradation Assay: In the same lysates, probe for p62, a selective autophagy receptor degraded along with its cargo. A successful and complete autophagic flux is indicated by an increase in LC3-II coupled with a decrease in p62 levels [55].
- Inhibition Controls: Co-treat cells with a known autophagy inhibitor such as chloroquine (which prevents lysosomal degradation). This should cause accumulation of both LC3-II and p62, validating that the observed effects are due to authentic autophagic activity.
The Scientist's Toolkit: Key Reagents and Materials
Successful TPD research and development relies on a suite of specialized reagents and tools. The table below details essential items for working with TPD therapeutics.
Table 3: Essential Research Reagent Solutions for TPD
| Reagent/Material | Function in TPD Research | Example Application |
|---|---|---|
| E3 Ligase Ligands | Recruit specific E3 ubiquitin ligases to form the ternary complex [67]. | VHL and CRBN ligands are widely used in PROTAC design and synthesis [67] [93]. |
| LC3 Antibodies | Detect and quantify LC3-I to LC3-II conversion, a key marker of autophagosome formation [55]. | Western blot analysis to confirm autophagy engagement by ATTECs or AUTACs [68]. |
| Ubiquitin-Activating Enzyme (E1) Inhibitor (e.g., TAK-243) | Serves as a negative control to block the UPS pathway upstream [92]. | Confirms that protein degradation by a PROTAC is ubiquitin-dependent. |
| Lysosomal Inhibitors (e.g., Chloroquine, Bafilomycin A1) | Block autophagic flux by preventing lysosomal acidification or fusion [68]. | Validates autophagy-dependent degradation in AUTAC or ATTEC assays. |
| Proteasome Activity Assay Kits | Measure chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasome [27]. | Assess if TPD treatment impacts overall proteasome function or capacity. |
| Nanocarriers (Liposomes, Polymeric NPs) | Improve the solubility, stability, and targeted delivery of TPD agents [43]. | In vivo delivery of degraders with poor pharmacokinetics to specific tissues. |
The strategic choice between UPS and autophagy-mediated TPD involves a critical trade-off between the well-established but more restricted UPS pathway and the broader substrate scope of the autophagy pathway, which presents its own unique delivery and selectivity challenges. The experimental data and protocols outlined herein provide a framework for the systematic, objective comparison of these emerging therapeutics. Overcoming the persistent hurdles of bioavailability and tissue-specific targeting—potentially through the innovative application of nanotechnology and advanced drug formulation [43]—will be paramount to fully realizing the clinical potential of TPD across a wide spectrum of human diseases.
Strategic Selection: A Comparative Analysis of UPS and Autophagy for Target Degradation
The faithful maintenance of cellular function requires precise management of the proteome, a process known as proteostasis. The ubiquitin-proteasome system (UPS) and autophagy-lysosomal system represent the two major pillars of cellular protein quality control, responsible for the selective degradation of dysfunctional, misfolded, or surplus proteins and organelles [95] [96]. While both systems utilize ubiquitin as a degradation signal, they exhibit distinct yet complementary substrate specificities, temporal dynamics, and functional capabilities. The UPS primarily targets soluble, short-lived proteins for rapid degradation, whereas autophagy specializes in the clearance of larger, more complex structures such as protein aggregates and entire organelles [27]. Understanding the precise scope and limitations of each pathway is fundamental to elucidating their roles in cellular homeostasis and their contributions to disease pathogenesis, particularly in neurodegenerative disorders characterized by pathological protein aggregation.
System Mechanisms and Substrate Specificity
The Ubiquitin-Proteasome System (UPS): Mechanism and Substrate Profile
The UPS is a highly selective, ATP-dependent proteolytic system responsible for the majority of targeted protein degradation in eukaryotic cells. Its mechanism involves a coordinated enzymatic cascade: an E1 ubiquitin-activating enzyme activates ubiquitin, which is then transferred to an E2 ubiquitin-conjugating enzyme, and finally, an E3 ubiquitin ligase confers substrate specificity by catalyzing the attachment of ubiquitin chains to target proteins [95] [27]. Polyubiquitinated substrates are recognized by the 26S proteasome, a multi-subunit complex comprising a 20S core particle with proteolytic activity and one or two 19S regulatory particles that facilitate substrate recognition, deubiquitination, and unfolding [27]. The unfolded polypeptide is then translocated into the proteolytic core for degradation into small peptides.
The UPS is exquisitely suited for the degradation of soluble, short-lived proteins, including cell cycle regulators, transcription factors, and signaling molecules [95]. It is also the primary system for handling soluble misfolded proteins that result from synthetic errors or damage [96]. A key structural limitation of the UPS is the narrow entrance pore of the 20S proteasome, which restricts the passage of large, oligomeric, or tightly folded structures. Consequently, the UPS is largely incapable of degrading insoluble protein aggregates or large macromolecular complexes without prior disaggregation or dissolution, a process that is often inefficient within the cell [97].
The Autophagy-Lysosomal System: Mechanism and Substrate Profile
Autophagy encompasses several pathways—macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA)—that deliver cargo to the lysosome for degradation. Macroautophagy (hereafter autophagy) involves the de novo formation of a double-membraned phagophore that expands to engulf cytoplasmic material, forming an autophagosome. This vesicle subsequently fuses with a lysosome to create an autolysosome, where the encapsulated contents are degraded by acidic hydrolases [96] [27]. This process is mediated by a suite of autophagy-related (ATG) proteins and two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L1 complex and the microtubule-associated protein 1 light chain 3 (LC3)-phosphatidylethanolamine conjugate [96] [27].
Unlike the UPS, autophagy can handle a vast and diverse array of cargo, primarily through selective forms of the pathway:
- Aggrephagy: Selective autophagy of protein aggregates, often initiated by the ubiquitination of the aggregate and recognition by autophagy receptors like p62/SQSTM1 [95] [98].
- Mitophagy: Selective autophagy of damaged or superfluous mitochondria, famously regulated by the PINK1-PARKIN pathway which marks depolarized mitochondria with ubiquitin chains for autophagic clearance [99].
- Chaperone-Mediated Autophagy (CMA): A selective process that directly translocates individual proteins bearing a KFERQ-like motif across the lysosomal membrane via the LAMP-2A receptor [96].
The autophagic system is uniquely equipped to degrade insoluble protein aggregates, dysfunctional organelles, and long-lived proteins [27]. Its capacity to engulf entire cellular structures makes it indispensable for cellular renovation and survival under stress conditions where the UPS is overwhelmed or ineffective.
Table 1: Comparative Overview of UPS and Autophagy Pathways
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosomal System |
|---|---|---|
| Primary Substrates | Soluble, short-lived proteins; soluble misfolded proteins [27] | Insoluble protein aggregates, organelles, long-lived proteins [27] |
| Degradation Machinery | 26S Proteasome (20S core + 19S cap) [27] | Lysosome [96] |
| Key Recognition Signal | Primarily K48-linked ubiquitin chains [27] | Ubiquitin (K63-linked, etc.); LC3-interacting regions (LIRs) in receptors [27] [100] |
| Selectivity Mediators | E3 Ubiquitin Ligases (e.g., PARKIN) [95] [99] | Autophagy Receptors (e.g., p62, Optineurin, Tollip) [98] [100] |
| Temporal Response | Rapid (minutes) [95] | Slower (hours) [95] |
| Energetic Cost | High (ATP-dependent unfolding) [27] | Lower (bulk engulfment) [96] |
Diagram 1: Comparative pathways of the UPS and autophagy-lysosomal system.
Direct Comparative Analysis of Substrate Degradation
The functional dichotomy between the UPS and autophagy becomes most apparent when examining their efficacy against different types of cellular cargo. The following table synthesizes experimental evidence regarding their respective capabilities.
Table 2: Substrate Degradation Efficacy of UPS vs. Autophagy
| Substrate Type | UPS Efficacy | Autophagy Efficacy | Key Experimental Evidence & Notes |
|---|---|---|---|
| Soluble Misfolded Proteins | High [27] | Moderate [27] | UPS is the primary degradation route; autophagy can engage via CMA or selective macroautophagy [96]. |
| Oligomers & Small Aggregates | Low [97] | High [97] [98] | UPS access is sterically hindered; autophagy receptors (e.g., Tollip) specifically target oligomers for degradation [98]. |
| Mature Amyloid Fibrils & Large Aggregates | Very Low / None [97] | High (Primary Pathway) [97] [68] | These large, insoluble structures are exclusive substrates for autophagic clearance [68]. |
| Damaged Organelles (e.g., Mitochondria) | Limited | High (Primary Pathway) [99] | UPS can degrade specific OMM proteins (e.g., via PARKIN) to prime mitophagy, but full organelle clearance requires autophagy [99]. |
| Long-lived Proteins | Low | High [27] | Autophagy is the major pathway for turnover of proteins with slow metabolic rates. |
The limitations of the UPS in handling aggregated proteins are a cornerstone of the pathophysiology of many neurodegenerative diseases. For instance, in Parkinson's disease, toxic aggregates of α-synuclein can overwhelm the UPS and must be cleared by autophagy [95]. Similarly, in Huntington's disease and amyotrophic lateral sclerosis (ALS), proteins with expanded polyglutamine tracts or TDP-43 aggregates, respectively, form inclusions that are predominantly degraded by autophagy [95] [98]. This compartmentalization of labor, however, is not absolute. Significant crosstalk exists between the two systems. Inhibition of the UPS can upregulate autophagy as a compensatory mechanism, and conversely, impaired autophagy can lead to proteasomal overload [99] [27]. Furthermore, for certain processes like mitophagy, the systems collaborate intimately: the UPS performs the initial "priming" of mitochondria by degrading specific outer membrane proteins, which then facilitates the efficient encapsulation and degradation of the entire organelle by autophagy [99].
Key Experimental Approaches and Methodologies
Investigating the roles and efficiencies of the UPS and autophagy requires a suite of well-established experimental protocols. Below are detailed methodologies for key assays used in the field.
Cycloheximide Chase Assay to Measure Protein Half-Life
This classic biochemical assay is used to determine the half-life of a protein and infer its primary degradation pathway.
Detailed Protocol [101]:
- Cell Preparation & Induction: Culture cells (e.g., yeast or mammalian cell lines) expressing the protein of interest under an inducible promoter (e.g., GAL1 promoter in yeast). Grow cells to mid-log phase.
- Translation Inhibition: Add a high concentration of cycloheximide (typically 50-100 µg/mL) to the culture medium. This inhibitor halts de novo protein synthesis, allowing researchers to track the fate of the existing pool of the protein.
- Time-Point Sampling: Collect aliquots of the cell culture at defined time points post-cycloheximide addition (e.g., 0, 15, 30, 60, 120 minutes).
- Cell Lysis and Solubilization: Rapidly pellet cells and lyse them. For yeast, mechanical disruption with glass beads in a lysis buffer containing detergents (e.g., NP-40) or chaotropic agents (e.g., 8M urea) is effective. Boiling in SDS sample buffer is a quicker alternative that denatures proteases, preventing post-lysis degradation [101].
- Protein Quantification: Resolve proteins by SDS-PAGE and perform Western blotting analysis using an antibody specific to the protein of interest.
- Data Analysis: Quantify the band intensity at each time point. Plot the relative protein level (logarithmic scale) against time. The half-life is the time taken for the protein signal to decrease by 50%. A shortened half-life upon proteasomal inhibition (e.g., with MG132) implicates the UPS, while a delay upon autophagy inhibition (e.g., with bafilomycin A1) points toward autophagic degradation.
Fluorescence Recovery After Photobleaching (FRAP) for Aggregate Dynamics
FRAP is a powerful live-cell imaging technique to study the dynamics and exchange of molecules within protein aggregates, informing on their solubility and engagement with quality control systems.
Detailed Protocol [102]:
- Sample Preparation: Transfert cells with a plasmid encoding the protein of interest (e.g., polyQ-expanded Huntingtin) fused to a fluorescent protein (e.g., GFP). Culture cells on glass-bottom dishes for 24-48 hours to allow for expression and potential aggregate formation.
- Microscope Configuration: Use a laser scanning confocal microscope equipped with a high-intensity laser (e.g., 488 nm argon laser for GFP) and a 40x or 63x oil-immersion objective. Set the laser power and detector gain to achieve a bright signal without saturation or excessive baseline photobleaching.
- Defining Regions of Interest (ROI): Select a cell with a visible fluorescent aggregate. Define two ROIs: one covering the aggregate to be bleached (bleach ROI) and another in a nearby cytosolic area for background correction.
- Photobleaching and Recovery: Acquire a few pre-bleach images to establish the baseline fluorescence. Illuminate the bleach ROI with a brief, high-intensity laser pulse to irreversibly bleach the fluorescent molecules within it. Immediately after bleaching, switch back to low-intensity laser scanning to acquire a time-lapse series of images to monitor the fluorescence recovery in the bleach ROI.
- Data Analysis: Normalize the fluorescence intensity in the bleach ROI to the pre-bleach value and the cytosolic background. Plot the normalized recovery over time to generate a FRAP curve. A rapid and complete recovery indicates high mobility and a dynamic, soluble-like state accessible to cellular machineries. A slow or incomplete recovery suggests a immobile, structured aggregate, characteristic of a substrate primarily for autophagic clearance [102].
Monitoring Ubiquitin Linkage Types on Misfolded Proteins
The type of ubiquitin chain attached to a substrate can determine its fate, with K48-linkages typically targeting to the proteasome and K63-linkages often signaling for autophagic degradation.
Detailed Protocol [101]:
- Immunoprecipitation: Lyse cells expressing the tagged misfolded protein under nondenaturing conditions (to preserve protein interactions). Incubate the lysate with an antibody specific to the tag of the misfolded protein and pull down the immune complexes with Protein A/G beads.
- Elution and Denaturation: Wash the beads thoroughly and elute the bound proteins under denaturing conditions (e.g., with SDS).
- Linkage-Specific Detection:
- Western Blotting: Separate the eluted proteins by SDS-PAGE and perform Western blotting. Instead of a general ubiquitin antibody, use linkage-specific ubiquitin antibodies (e.g., anti-K48-linkage or anti-K63-linkage specific antibodies) to determine the predominant chain type conjugated to the substrate.
- ELISA-based Quantification: As a more quantitative approach, the immunoprecipitated material can be immobilized on a 96-well plate. The plate is then probed with linkage-specific ubiquitin antibodies in a standard ELISA protocol, allowing for high-throughput quantification of specific ubiquitin linkages [101].
Diagram 2: Experimental workflow for analyzing misfolded protein degradation pathways.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues critical reagents and their applications for studying UPS and autophagy.
Table 3: Key Reagents for Protein Degradation Research
| Reagent / Tool | Category | Primary Function & Application |
|---|---|---|
| MG132 / Bortezomib | Pharmacological Inhibitor | Potent and reversible inhibitors of the proteasome's chymotrypsin-like activity. Used to block UPS function and assess its contribution to protein turnover [27]. |
| Bafilomycin A1 | Pharmacological Inhibitor | Inhibits the vacuolar-type H+-ATPase (V-ATPase), preventing lysosomal acidification and thus blocking autophagic flux at the degradation stage [27]. |
| Chloroquine | Pharmacological Inhibitor | A lysosomotropic agent that raises lysosomal pH, impairing hydrolase activity and inhibiting autophagic degradation. |
| 3-Methyladenine (3-MA) | Pharmacological Inhibitor | A Class III PI3K inhibitor that blocks autophagosome formation by inhibiting the VPS34 complex during the nucleation stage. |
| Cycloheximide | Protein Synthesis Inhibitor | Used in chase assays to halt new protein synthesis, allowing measurement of the degradation rate of existing proteins [101]. |
| LC3B Antibody | Biological Reagent | A key biomarker for autophagy. Detects both cytosolic LC3-I and lipidated, autophagosome-associated LC3-II by Western blot. Punctate LC3 staining by immunofluorescence indicates autophagosome formation. |
| p62/SQSTM1 Antibody | Biological Reagent | Detects the selective autophagy receptor p62. Its accumulation often indicates blocked autophagic flux, while its degradation correlates with active autophagy. |
| Linkage-Specific Ubiquitin Antibodies | Biological Reagent | Antibodies specific for K48, K63, or other ubiquitin linkages are essential for determining the fate of ubiquitylated substrates via Western blot or ELISA [101]. |
| PARKIN / PINK1 Expression Constructs | Genetic Tool | Used to induce and study mitophagy in cellular models. PARKIN is an E3 ubiquitin ligase recruited to depolarized mitochondria by PINK1 kinase [99]. |
| Tollip / Cue5 Expression Constructs | Genetic Tool | Key adaptor proteins for aggrephagy. Their overexpression can enhance aggregate clearance, while knockdown impairs it, providing a tool to manipulate this pathway [98]. |
The UPS and autophagy operate as a coordinated, integrated network to maintain proteostasis, with their substrate scope defined by fundamental physicochemical and structural limitations. The UPS excels in the rapid, selective degradation of soluble proteins but is sterically excluded from handling large aggregates and organelles. Autophagy serves as the ultimate clearance mechanism for these more complex and bulky cargoes. The experimental dissection of these pathways relies on a combination of biochemical, imaging, and genetic tools that allow researchers to interrogate protein half-life, complex dynamics, and degradation signals. Understanding these specialized roles and their interplay is not only critical for basic cell biology but also for developing targeted therapeutic strategies for neurodegenerative diseases and cancer, where modulating the activity of these pathways holds significant promise.
The ubiquitin-proteasome system (UPS) and autophagy are the two primary degradation pathways responsible for maintaining cellular proteostasis. The ubiquitin-proteasome system (UPS) is highly selective, targeting primarily short-lived, soluble, and misfolded proteins for rapid degradation [36] [1]. In contrast, autophagy handles the clearance of bulk cytoplasmic material, including long-lived proteins, insoluble protein aggregates, and damaged organelles [76] [36]. Understanding the distinct kinetic profiles and efficiency parameters of these pathways is crucial for researchers developing targeted protein degradation therapies, especially for diseases involving misfolded proteins, such as neurodegenerative disorders [103] [104]. This guide provides a direct, data-driven comparison of pathway throughput to inform experimental design and degrader optimization.
Quantitative Comparison of Degradation Parameters
The following tables summarize key quantitative parameters that define the throughput and operational characteristics of the UPS and autophagy pathways.
Table 1: Kinetic and Efficiency Parameters of UPS vs. Autophagy
| Parameter | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome Pathway |
|---|---|---|
| Primary Substrate Types | Short-lived, soluble misfolded proteins [36] [1] | Long-lived proteins, protein aggregates, damaged organelles [55] [36] |
| Degradation Rate | Rapid (minutes to a few hours); t¹/₂ of short-lived proteins can be ≤2 hours [105] | Slower (hours); gradual decrease over time [105] |
| Maximal Degradation (Dmax) | Can achieve >90% target knockdown with effective degraders (e.g., PROTACs) [105] | Can be partial (30-60% target remains) or complete, depending on strategy and cargo [105] |
| Catalytic Nature | Event-driven; one degrader can mediate multiple degradation events [105] | Event-driven; degraders can act catalytically [55] |
| Key Structural Constraint | Proteasome barrel pore limits substrate size [1] | No strict size limit; can degrade large aggregates and organelles [55] |
Table 2: Key Degrader Technologies and Their Performance
| Degrader Technology | Primary Pathway | Typical Targets | Key Kinetic Considerations |
|---|---|---|---|
| PROTAC [105] [36] | UPS | Intracellular proteins (e.g., kinases, transcription factors) | Rapid, dose-dependent degradation possible; can exhibit "hook effect" at high concentrations [105] |
| Molecular Glue [105] [36] | UPS | Intracellular proteins, often difficult targets | Smaller size can improve bioavailability; degradation kinetics can be rapid [105] |
| ATTEC/AUTAC [55] | Autophagy | Proteins, protein aggregates, organelles | Leverages selective autophagy; useful for targets resistant to UPS degradation [55] |
| LYTAC [105] | Lysosome (Endocytosis) | Extracellular and membrane proteins | Efficiency depends on cell surface receptor expression levels [55] [105] |
Experimental Protocols for Kinetic Profiling
Accurately measuring the kinetics of targeted protein degradation is essential for comparing pathway throughput and optimizing degrader compounds. The following protocols outline key methodologies.
Time-Course Experiments for Degradation Monitoring
Function: To measure the rate of target protein loss and subsequent recovery over time in degrader-treated cells [105]. Procedure:
- Cell Treatment: Apply the degrader compound (e.g., PROTAC, AUTAC) at the desired concentration to cells. A DMSO vehicle control is mandatory.
- Sample Collection: Harvest cell lysates at multiple time points post-treatment (e.g., 0.5, 1, 2, 4, 8, 24 hours) [105].
- Target Quantification: Analyze target protein levels in lysates using Western blot or immunoassays (e.g., ELISA) [105].
- Data Analysis: Plot the percentage of remaining target protein versus time to generate a degradation curve. From this curve, key kinetic parameters like degradation half-life (t¹/₂), Dmax, and recovery rate can be derived [105].
Real-Time Live-Cell Assays
Function: To monitor protein degradation kinetics continuously in live cells, providing high-temporal-resolution data [105]. Procedure:
- Reporter Engineering: Tag the endogenous target protein gene with a luminescent (e.g., HiBiT, NanoLuc) or fluorescent reporter [105].
- Signal Measurement: Seed engineered cells into a multi-well plate and treat with the degrader. For luminescent reporters, add substrate to the media and place the plate in a reader that maintains physiological conditions.
- Kinetic Recording: Measure the luminescence/fluorescence signal at frequent intervals (e.g., every 15-30 minutes) over 24-72 hours. As the target protein is degraded, the signal diminishes [105].
- Data Processing: Plot the signal intensity over time to obtain a real-time degradation profile, allowing for precise calculation of degradation onset and rate.
Pulse-Chase and Turnover Analysis
Function: To isolate the degradation rate from ongoing protein synthesis and measure the intrinsic half-life of a protein [105]. Procedure:
- Pulse Phase: Briefly incubate cells with labeled amino acids (e.g., ³⁵S-methionine) to incorporate the label into newly synthesized proteins.
- Chase Phase: Replace the labeled medium with a large excess of unlabeled amino acids. This "chases" the label into the existing protein pool and prevents new incorporation.
- Degrader Application: Add the degrader compound at the start of the chase phase or at a later point.
- Sampling and Analysis: Collect cell samples at various time points during the chase. Immunoprecipitate the target protein and measure the amount of remaining labeled protein. The decay curve reveals the protein's half-life and the degrader's effect on its clearance rate [105].
Signaling Pathways and System Interplay
The UPS and autophagy are not independent but form a coordinated network for maintaining proteostasis. The diagram below illustrates their key components and functional crosstalk.
Diagram 1: UPS and Autophagy Pathways in Protein Degradation. This diagram illustrates the distinct fates of a ubiquitinated protein. K48-linked chains typically target substrates for rapid proteasomal degradation, while K63-linked chains and other signals can be recognized by adaptor proteins like p62, which shuttle cargo to the autophagosome via LC3 for lysosomal degradation. Dashed lines indicate documented crosstalk and compensatory interplay between the two systems [36] [104] [1].
The Scientist's Toolkit: Essential Research Reagents
Successful profiling of degradation kinetics requires a suite of reliable tools and reagents. The following table catalogues essential solutions for researchers in this field.
Table 3: Key Research Reagent Solutions for Degradation Studies
| Reagent / Tool | Function/Description | Primary Application |
|---|---|---|
| HiBiT / NanoLuc Tagging [105] | A small luciferase tag that provides a highly sensitive, quantitative luminescent signal for low-abundance proteins in live cells. | Real-time, live-cell kinetic assays for monitoring protein degradation [105]. |
| LC3 Antibodies | Specific antibodies against LC3-I and lipidated LC3-II forms, which serve as a key marker for autophagosome formation and flux [55]. | Western blot analysis to monitor autophagy induction and progression in response to degraders [55]. |
| p62/SQSTM1 Antibodies | Antibodies against the selective autophagy receptor p62, whose levels inversely correlate with autophagic flux when LC3-II is elevated [55]. | Assessing autophagic flux and selectivity in degradation experiments [55]. |
| Proteasome Inhibitors(e.g., MG132, Bortezomib) | Small molecules that specifically inhibit the proteolytic activity of the 26S proteasome [103]. | Validating UPS-specific degradation; studying UPS-autophagy crosstalk by blocking the proteasome [103]. |
| Autophagy Inhibitors(e.g., Bafilomycin A1, Chloroquine) | Compounds that prevent autophagosome-lysosome fusion or lysosomal acidification, blocking the final stage of autophagy [55]. | Validating autophagy-mediated degradation; measuring autophagic flux. |
| Ubiquitin Remnant Profiling Kits | Tools (e.g., using UbiScan technology) to enrich and profile ubiquitinated peptides from cell lysates. | Mechanistic studies to detect target ubiquitination, an early step that often precedes degradation [105]. |
The UPS and autophagy pathways offer complementary yet distinct advantages for targeted protein degradation. The UPS excels in speed and selectivity for soluble targets, while autophagy provides unique capacity for handling large, complex aggregates. Critical to therapeutic development is the recognition of their profound interplay and crosstalk, where inhibition of one system can upregulate the other as a compensatory mechanism [103] [1]. A thorough kinetic profile—encompassing degradation rate, maximal degradation, and recovery dynamics—is therefore indispensable for selecting the optimal degradation strategy and designing effective degrader molecules for research and therapeutic applications [105].
In the evolving landscape of targeted protein degradation, two principal cellular pathways—the ubiquitin-proteasome system (UPS) and autophagy—offer complementary mechanisms for eliminating disease-causing proteins. The ubiquitin-proteasome system primarily degrades short-lived proteins and soluble misfolded proteins through a sophisticated enzymatic cascade that tags substrates with ubiquitin for proteasomal recognition and destruction [36]. In contrast, autophagy is a lysosome-dependent process that eliminates long-lived proteins, insoluble protein aggregates, and damaged organelles through encapsulation in double-membraned vesicles that fuse with lysosomes [106] [36]. For researchers and drug development professionals, the strategic selection between these pathways depends critically on the biophysical properties of the target protein, the nature of the pathology, and the desired therapeutic outcome. This guide provides a structured comparison of these degradation pathways, supported by experimental data and methodologies, to inform therapeutic development decisions for protein misfolding disorders.
Pathway Mechanisms and Molecular Players
The Ubiquitin-Proteasome System (UPS)
The UPS employs a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes to tag target proteins with polyubiquitin chains, primarily through K48 linkages, marking them for degradation by the 26S proteasome [36]. The E3 ubiquitin ligases provide substrate specificity, with over 600 encoded in the human genome, though current targeted protein degradation technologies primarily utilize a limited set including CRBN, VHL, MDM2, and IAPs [67]. This pathway is particularly efficient for degrading soluble, properly folded proteins and some misfolded proteins that haven't yet formed larger aggregates.
Table 1: Key UPS Components in Targeted Degradation
| Component | Role in UPS | Research Applications |
|---|---|---|
| E3 Ligases (CRBN, VHL) | Provide substrate specificity for ubiquitination | Recruited by PROTACs for targeted protein degradation [67] |
| Proteasome | 26S multicatalytic protease complex | Degrades ubiquitin-tagged proteins; inhibited for mechanistic studies [36] |
| Deubiquitinases (DUBs) | Remove ubiquitin chains | Validate ubiquitin-dependent degradation; potential therapeutic targets [67] |
| Ubiquitin | 76-amino acid protein tag | tagging system for degradation; used in pull-down assays [36] |
The Autophagy-Lysosomal Pathway
Autophagy involves the formation of double-membraned autophagosomes that engulf cellular components and deliver them to lysosomes for degradation. This process is orchestrated by evolutionarily conserved autophagy-related (ATG) proteins and can be activated by various cellular stresses, including nutrient deprivation and endoplasmic reticulum (ER) stress [106] [107]. The pathway is particularly suited for eliminating protein aggregates, damaged organelles, and long-lived proteins that are refractory to proteasomal degradation. Key regulators include the ULK/ATG13/FIP200 complex for induction, class III PI3K complex for vesicle nucleation, and LC3 conjugation systems for autophagosomal elongation [106].
Table 2: Key Autophagy Components and Research Tools
| Component | Role in Autophagy | Research Applications |
|---|---|---|
| LC3-I/LC3-II | Autophagosome membrane marker | Western blot and immunofluorescence to monitor autophagic flux [106] |
| p62/SQSTM1 | Autophagy adaptor protein | Degradation indicates functional autophagy; accumulates when impaired [106] |
| ULK1/2 complex | Initiates autophagosome formation | Target for autophagy induction/inhibition [106] |
| Beclin-1 | Part of PI3K complex for vesicle nucleation | Modulates autophagic activity; interacts with Bcl-2 [106] |
| LAMP1/2 | Lysosomal membrane proteins | Marker for lysosomal abundance and function [106] |
Matching Pathology to Degradation Pathway
Quantitative Comparison of Pathway Characteristics
Table 3: Strategic Selection Guide: UPS vs. Autophagy
| Parameter | Ubiquitin-Proteasome System | Autophagy-Lysosomal Pathway |
|---|---|---|
| Primary Substrates | Short-lived soluble proteins, properly folded proteins, some misfolded proteins [36] | Protein aggregates, damaged organelles, long-lived proteins, large complexes [106] [36] |
| Degradation Capacity | Individual proteins, requires unfolding | Bulk degradation, entire organelles, protein aggregates [106] |
| Therapeutic Approaches | PROTACs, Molecular Glues, dTAG, SNIPERs [67] [36] | AUTOTAC, LYTAC, small molecule inducers [108] |
| Ideal Pathologies | Oncology targets (kinases, nuclear receptors), some neurodegenerative targets [67] [109] | Aggregate-based diseases (ALS, Alzheimer's), organelle quality control [110] [107] |
| Key Advantages | High specificity, catalytic action, targets "undruggable" proteins [36] | Handles large aggregates, complements UPR, metabolic flexibility [106] [107] |
| Experimental Readouts | Decreased target protein by Western, increased polyubiquitination, cycloheximide chase [110] | LC3-I to LC3-II conversion, p62 degradation, fluorescence microscopy [106] [83] |
Pathway Selection for Specific Proteinopathies
UPS-Targetable Pathologies
The UPS is particularly effective for degrading soluble misfolded proteins before they form larger aggregates. Recent research demonstrates the potential of BioPROTACs for selectively targeting misfolded species while sparing native proteins. In a groundbreaking study targeting amyotrophic lateral sclerosis (ALS)-linked SOD1 variants, researchers developed a BioPROTAC termed "MisfoldUbL" that specifically degraded misfolded SOD1 while preserving natively folded protein [110]. This approach reduced insoluble SOD1 aggregates by up to 76% in cellular models and delayed disease progression in SOD1G93A transgenic mice, demonstrating the therapeutic potential of UPS-mediated degradation for specific misfolded proteins [110].
The UPS also shows promise for entanglement-type misfolding, a recently identified class of protein misfolding where sections of amino acids form improper loops or fail to form necessary entanglements. Atomic-scale simulations confirm these misfolds can be stable and evade cellular quality control systems, making them promising targets for UPS-mediated degradation [111].
Autophagy-Targetable Pathologies
Autophagy excels at degrading large protein aggregates and handling organelle quality control. In non-alcoholic fatty liver disease (NAFLD), autophagy helps clear lipid droplets and protein aggregates that accumulate during ER stress, with studies showing that autophagy induction can reduce hepatic steatosis by mitigating ER stress [107]. The pathway is particularly valuable when the UPS becomes overloaded, as demonstrated in cerebral ischemia models where a molecular switch from UPS to autophagy activation occurs during prolonged stress conditions [83].
Research has revealed specialized forms of autophagy, including ER-phagy (ER-specific autophagy), which counterbalances ER expansion during the unfolded protein response. In yeast models, ER volume increased more than 5-fold under UPR-inducing conditions, accompanied by formation of autophagosome-like structures specifically packed with membrane stacks from the expanded ER [112]. This selective ER sequestration required several autophagy genes and was essential for cell survival under severe ER stress, indicating a dedicated quality control mechanism for organelle homeostasis.
Experimental Approaches and Methodologies
Protocol: Assessing UPS Function and Target Engagement
Objective: Evaluate UPS-mediated degradation of a target protein using PROTAC treatment.
Methodology:
- Cell Treatment: Apply PROTAC compound (e.g., ARV-471 for estrogen receptor, ARV-110 for androgen receptor) at varying concentrations (typically 1 nM-10 μM) and time points (1-24 hours) to relevant cell lines [109].
- Inhibition Controls: Co-treat with proteasome inhibitor (MG132, 10-20 μM for 6 hours) or E1 ubiquitin-activating enzyme inhibitor (MLN7243, 1 μM for 12 hours) to confirm UPS dependence [110].
- Sample Collection: Harvest cells for Western blotting at predetermined time points.
- Western Blot Analysis:
- Probe for target protein to quantify degradation
- Assess polyubiquitinated proteins using ubiquitin-specific antibodies
- Monitor proteasome activity with proteasome subunit antibodies
- Include loading controls (GAPDH, actin) [110]
- Cycloheximide Chase: Treat cells with protein synthesis inhibitor cycloheximide (100 μg/mL) alongside PROTAC to measure protein half-life independently of new synthesis [110].
- Immunoprecipitation: Confirm ternary complex formation by immunoprecipitating the target protein and probing for associated E3 ligase.
Expected Outcomes: Dose- and time-dependent reduction in target protein levels that is blocked by proteasome inhibition, confirming UPS-mediated degradation.
Protocol: Monitoring Autophagic Flux
Objective: Quantify autophagic activity and lysosomal degradation in cellular models.
Methodology:
- Indicator Treatments:
- Induce autophagy with starvation (EBSS medium) or mTOR inhibitor (rapamycin, 100 nM for 4-24 hours)
- Inhibit autophagosome-lysosome fusion with bafilomycin A1 (100 nM for 4-6 hours) [106]
- Western Blot Analysis:
- Immunofluorescence Microscopy:
- Transfert cells with GFP-LC3 plasmid or use LC3 antibodies
- Quantify GFP-LC3 puncta per cell, representing autophagosomes [112]
- Co-stain with LAMP1/2 antibodies to visualize lysosomes and assess colocalization
- Transmission Electron Microscopy:
- Fix cells with glutaraldehyde and prepare thin sections
- Identify and quantify autophagic structures (autophagosomes, autolysosomes) per cell section [112]
- Long-Lived Protein Degradation Assay:
- Label proteins with radioactive [³H]leucine during 24-48 hour incubation
- Chase with cold leucine for 4-6 hours
- Measure trichloroacetic acid-soluble radioactivity in medium to quantify protein degradation [106]
Expected Outcomes: Increased LC3-II and decreased p62 under induction conditions; LC3-II accumulation with bafilomycin A1 treatment indicates functional autophagic flux.
Protocol: Evaluating the UPS-to-Autophagy Switch
Objective: Investigate the transition from UPS to autophagy under proteostatic stress.
Methodology:
- Stress Induction: Apply progressively severe stress (e.g., cerebral ischemia models with 10-60 min tMCAO, proteasome inhibitors, or expression of aggregation-prone proteins) [83].
- Temporal Analysis: Collect samples at multiple time points (0.5-72 hours post-stress) to capture dynamic changes.
- Simultaneous UPS/Autophagy Assessment:
- UPS Markers: Monitor ubiquitin conjugates, proteasome activity, BAG1 levels
- Autophagy Markers: Track LC3-I/II conversion, p62 degradation, BAG3 induction [83]
- Ratio Determination: Calculate BAG1/BAG3 ratio, where decreased ratio indicates shift toward autophagy [83].
- HDAC6 Monitoring: Assess HDAC6 levels, which increase during autophagy activation to facilitate aggresome formation and autophagic clearance [83].
- Immunofluorescence Colocalization: Perform double staining for ubiquitin with p62 or HSP70 to visualize protein aggregate formation and targeting to autophagy.
Expected Outcomes: Initial increase in ubiquitinated proteins followed by LC3-II induction and p62 decrease, with declining BAG1/BAG3 ratio indicating pathway switching.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Protein Degradation Studies
| Reagent/Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| UPS Modulators | MG132, Bortezomib, MLN7243 | Proteasome/E1 inhibition to confirm UPS dependence [110] | Blocks final degradation step; accumulates ubiquitinated substrates |
| Autophagy Modulators | Bafilomycin A1, Chloroquine, Rapamycin | Inhibit or induce autophagy to measure flux [106] | Bafilomycin blocks lysosomal fusion; rapamycin induces via mTOR inhibition |
| Pathway Reporters | GFP-LC3, Ubiquitin sensors, H2B-p62 | Live-cell imaging of pathway activity [112] | Visualize autophagosome formation, protein aggregation in real-time |
| PROTAC Molecules | ARV-471 (ER), ARV-110 (AR), CFT7455 (IKZF1/3) | Targeted protein degradation tools [109] | Demonstrate UPS-mediated degradation of specific targets |
| Antibody Panels | Anti-LC3, anti-p62, anti-ubiquitin, anti-BAG3 | Western blot, immunofluorescence readouts [83] | Quantify pathway activation and substrate clearance |
| Cellular Stressors | Tunicamycin, Thapsigargin, DTT, Cerebral ischemia models | Induce ER stress and UPR activation [111] [112] | Activate quality control pathways for mechanistic studies |
The strategic matching of target pathology to degradation pathway requires careful consideration of target properties, disease stage, and pathway capabilities. The UPS offers precision for soluble targets and early-stage pathologies, while autophagy provides capacity for aggregated proteins and advanced disease states. Emerging evidence of a regulated switch between these pathways under proteostatic stress [83] suggests potential combination approaches that sequentially or simultaneously engage both systems. As targeted degradation technologies advance, the integration of pathway knowledge with therapeutic design will enable more effective strategies for addressing protein misfolding diseases across therapeutic areas.
The maintenance of proteostasis is a critical cellular function, and the degradation of misfolded proteins is a central pillar of this process. The two primary cellular systems tasked with this function are the Ubiquitin-Proteasome System (UPS) and Autophagy. The UPS is renowned for its rapid, selective degradation of short-lived and soluble proteins, while the autophagy-lysosome pathway specializes in clearing bulky cargoes, including protein aggregates and damaged organelles. In the context of drug discovery, particularly for neurodegenerative diseases and cancer, understanding the strategic advantages and limitations of each pathway is paramount. This guide provides an objective comparison of these two systems, framing them within the modern paradigm of targeted degradation technologies.
Systematic Comparison of UPS and Autophagy
The following tables summarize the core characteristics, performance data, and therapeutic applicability of the UPS and autophagy pathways.
Table 1: Fundamental Characteristics and Degradation Performance
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome Pathway |
|---|---|---|
| Core Mechanism | Ubiquitin-tagged proteins degraded by the 26S proteasome [113] | Cargo sequestered in autophagosomes & degraded by lysosomal hydrolases [55] [114] |
| Primary Degradation Cargo | Short-lived, soluble, ubiquitinated proteins [113] | Protein aggregates, damaged organelles, long-lived proteins, intracellular pathogens [55] [115] |
| Degradation Capacity | Limited pore size; inefficient for bulk aggregates [55] | High capacity for large structures and bulk cytoplasm [55] |
| Key Degradation Signal | Ubiquitin chain [113] | Ubiquitin (for some selective types) or specific motifs (e.g., KFERQ for CMA) [55] |
| Selectivity | High (via E3 ubiquitin ligases) [113] | Can be non-selective (bulk) or highly selective (receptor-mediated) [55] |
| Representative Targeted Degradation Technology | PROTACs (Proteolysis-Targeting Chimeras) [55] | ATTEC, AUTAC, AUTOTAC, LYTAC [55] |
Table 2: Therapeutic Applicability and Strategic Considerations
| Aspect | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome Pathway |
|---|---|---|
| Therapeutic Context | Oncology (e.g., ARV-110 targeting AR in Phase II) [55] | Neurodegenerative diseases (e.g., Huntington's, Alzheimer's), Oncology [55] [114] |
| Key Advantages | High specificity, rapid turnover, covers ~80% of intracellular protein degradation, well-suited for transcription factors & non-enzymatic proteins [55] | Broad substrate scope (proteins to organelles), can degrade "undruggable" aggregates, does not rely solely on ubiquitination [55] |
| Major Drawbacks | Inefficient against large protein aggregates; proteasome dysfunction can produce cytotoxic products [55] | Generally slower than UPS; complex regulation and contextual dual roles in diseases like cancer [115] [114] |
| Limitations of Derived Technologies | PROTACs are inefficient against aggregates and depend on specific E3 ligase expression [55] | Technologies like LYTAC depend on variable cell surface receptor expression [55] |
Experimental Insights and Methodologies
Key Experimental Models for Pathway Interrogation
Research into the UPS and autophagy often relies on specific, quantifiable experimental models.
- For UPS and Mitophagy: A common protocol involves inducing mitochondrial stress in cell lines (e.g., HEK293T) using compounds like CCCP (a protonophore) or Staurosporine. The recruitment of key proteins like the E3 ligase PARK2 (PARKIN) and the autophagy protein ATG5 to mitochondria is then monitored via co-immunoprecipitation (Co-IP) and Western blotting to study functional crosstalk [113].
- For Autophagic Flux: A standard methodology measures "autophagic flux" by tracking the turnover of LC3, a canonical autophagy marker. This is typically done via Western blot to monitor the lipidated form (LC3-II) levels in the presence and absence of lysosomal inhibitors (e.g., chloroquine or bafilomycin A1). A concurrent measurement of p62/SQSTM1 levels is used, where increased LC3-II with decreased p62 indicates unimpeded flux [55].
Visualizing the Crosstalk: UPS and Autophagy in Mitophagy
The following diagram, generated using Graphviz DOT language, illustrates the complex, collaborative interplay between UPS and autophagy components during the critical quality control process of mitophagy, as revealed by recent research [113].
Diagram 1: UPS-Autophagy Crosstalk in Mitophagy.
Core Signaling and Regulatory Pathways
The decision to activate autophagy is governed by sophisticated nutrient and energy-sensing pathways. The following diagram depicts the core regulatory network that controls autophagy initiation, integrating signals from key kinases and metabolic sensors [115] [114] [116].
Diagram 2: Core Regulation of Autophagy Initiation.
The Scientist's Toolkit: Key Research Reagents
This table catalogs essential reagents and tools for investigating UPS and autophagy pathways, drawing from methodologies cited in the literature.
Table 3: Key Research Reagent Solutions for UPS and Autophagy Studies
| Reagent / Tool | Function / Target | Experimental Application |
|---|---|---|
| CCCP (Carbonyl cyanide m-chlorophenyl hydrazone) | Mitochondrial uncoupler | Induces mitochondrial depolarization to study mitophagy [113] |
| Bafilomycin A1 | V-ATPase inhibitor (blocks lysosomal acidification) | Used in flux assays to inhibit autophagosome degradation, allowing LC3-II accumulation measurement [55] |
| Anti-LC3 Antibody | Recognizes LC3-I (cytosolic) and LC3-II (lipidated, autophagosome-bound) | Western blotting, immunofluorescence to monitor autophagosome number and autophagic activity [55] |
| Anti-p62/SQSTM1 Antibody | Detects selective autophagy receptor that links ubiquitinated cargo to LC3 | Western blotting; levels inversely correlate with autophagic flux when autophagy is induced [55] |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Inhibit chymotrypsin-like activity of the 20S proteasome | To study UPS function, protein half-life, and the effects of proteasome inhibition on alternative pathways like autophagy [113] |
| siRNA/shRNA vs. ATG5, PSMA7 | Gene knockdown of core autophagy (ATG5) and proteasome (PSMA7) components | To validate the functional requirement of specific proteins in degradation pathways and study pathway crosstalk [113] |
The ubiquitin-proteasome system (UPS) and autophagy represent two fundamental pillars of cellular protein quality control. The UPS is a selective proteolytic system responsible for the rapid degradation of short-lived regulatory proteins and soluble misfolded proteins, typically tagged with K48-linked ubiquitin chains for processive degradation by the proteasome [10] [117]. In contrast, autophagy, particularly macroautophagy, functions as a bulk degradative system that clears protein aggregates, damaged organelles, and other cytoplasmic constituents via lysosomal hydrolases [10] [55]. While historically viewed as independent pathways, emerging research reveals extensive crosstalk and functional cooperation between these systems, forming a single proteolytic network that maintains cellular homeostasis under varying stress conditions [117]. This comparative guide examines the distinct characteristics, experimental methodologies, and therapeutic applications of both pathways, with particular focus on emerging technologies that leverage their integration for targeted protein degradation.
System Mechanisms and Comparative Analysis
Molecular Machinery and Degradation Signals
The UPS operates through a coordinated enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that conjugate ubiquitin to target proteins [10]. E3 ubiquitin ligases, including HECT, RING, U-box, and RBR types, provide substrate specificity [118]. Polyubiquitination via K48 linkages serves as the primary proteasomal degron, recognized by proteasome-associated adaptors like RPN10 and RPN13 [117]. The 26S proteasome, composed of a 20S core protease and 19S regulatory particle, then degrades the tagged substrates [118].
Autophagy involves the formation of double-membrane autophagosomes that engulf cytoplasmic cargo. Key autophagy-related proteins (ATGs) regulate this process, with LC3/ATG8 serving as a critical marker for autophagosome formation [55]. Selective autophagy receptors like p62/SQSTM1 and NBR1 bridge ubiquitinated cargo to LC3 via LC3-interacting regions (LIRs), with K63-linked ubiquitin chains often serving as autophagic degrons [117] [55]. Chaperone-mediated autophagy (CMA) provides an alternative route through direct translocation of substrates bearing KFERQ-like motifs across the lysosomal membrane via LAMP2A receptors [55].
Table 1: Comparative Features of UPS and Autophagy Pathways
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy-Lysosome System |
|---|---|---|
| Primary Function | Selective degradation of short-lived proteins | Bulk degradation of aggregates, organelles |
| Degradation Signal | K48-ubiquitin chains (primary) | K63-ubiquitin chains, receptor-mediated |
| Key Enzymes/Components | E1-E2-E3 enzyme cascade, 26S proteasome | ATG proteins, LC3, p62, LAMP2A (CMA) |
| Energy Dependency | ATP-dependent | ATP-dependent |
| Degradation Capacity | Individual proteins | Protein aggregates, organelles |
| Therapeutic Technologies | PROTACs | ATTECs, AUTACs, AUTOTAC, LYTACs |
| Substrate Examples | Cyclin, p53, misfolded ER proteins | Protein aggregates, damaged mitochondria |
Pathway Crosstalk and Compensatory Regulation
Cells operate a dynamic equilibrium between UPS and autophagy, with compensatory upregulation of one pathway when the other is impaired [117]. Under proteotoxic stress, ubiquitinated proteins resistant to proteasomal degradation are often redirected to autophagy via receptors like p62 and HDAC6, which binds ubiquitin chains and facilitates aggresome formation for autophagic clearance [10] [117]. This coordination is particularly evident in protein quality control, where misfolded proteins are first ubiquitinated by E3 ligases like CHIP, then directed to either pathway based on the nature of ubiquitin chains and cellular conditions [10]. The ubiquitin code thus serves as a common language interpreted by both degradation systems, with chain topology determining destination [117].
Experimental Analysis of Pathway Performance
Methodologies for Assessing Degradation Efficiency
UPS Activity assays typically employ reporter substrates like ubiquitin-fusion degradation (UFD) pathway targets or naturally short-lived proteins (e.g., p53) tagged with fluorescent proteins or luciferase [118]. Experimental protocols involve:
- Cycloheximide Chase Analysis: Cells treated with translation inhibitor cycloheximide, with samples collected over time to monitor protein decay via immunoblotting [119]
- Proteasome Inhibition: MG132 or bortezomib treatment to validate UPS-dependent degradation, observing substrate stabilization [120] [119]
- Ubiquitination Status: Co-immunoprecipitation under denaturing conditions to detect polyubiquitinated species [119]
Autophagy Flux Measurement employs multiple complementary approaches:
- LC3-I to LC3-II Conversion: Immunoblot detection of lipidated LC3-II, which correlates with autophagosome number [55]
- p62/SQSTM1 Degradation Assay: Monitoring p62 levels, as reduced p62 with elevated LC3-II indicates functional autophagy flux [55]
- Tandem Fluorescent LC3 Reporter: Using mRFP-GFP-LC3 to distinguish autophagosomes (yellow puncta) from autolysosomes (red puncta) based on GFP quenching in acidic compartments [55]
Table 2: Experimental Approaches for Pathway Analysis
| Parameter Assessed | UPS-Focused Methods | Autophagy-Focused Methods |
|---|---|---|
| Degradation Kinetics | Cycloheximide chase, proteasome inhibitors | Bafilomycin A1 treatment, lysosomal inhibitors |
| Substrate Modification | Ubiquitination assays, chain linkage analysis | Phosphorylation status, receptor interactions |
| Pathway Activity | Proteasome activity probes, reporter substrates | LC3 flux analysis, p62 degradation kinetics |
| Morphological Assessment | Immunofluorescence for ubiquitin aggregates | Fluorescent reporter constructs (mRFP-GFP-LC3) |
| Functional Compensation | Dual inhibition studies, stress response markers | Sequential pathway inhibition, aggregation analysis |
Performance Metrics in Disease Contexts
In neurodegenerative diseases characterized by protein aggregation (e.g., Alzheimer's, Huntington's), autophagy demonstrates superior capacity for eliminating large, insoluble aggregates that resist proteasomal degradation [10] [55]. Quantitative studies show autophagy can clear protein aggregates 3-5 times more efficiently than UPS in these contexts [55]. Conversely, for rapid degradation of regulatory proteins like cell cycle controllers and transcription factors, UPS exhibits faster kinetics, typically completing degradation within hours versus the slower autophagy process which may require 12-24 hours for significant cargo clearance [10] [117].
The integration of both pathways is exemplified in aggresome processing: UPS components initially ubiquitinate misfolded proteins, which are then sequestered into aggressomes via HDAC6 and finally cleared by autophagy [117]. This sequential cooperation highlights the functional specialization of each system while demonstrating their synergistic potential in maintaining proteostasis.
Emerging Integrated Degradation Technologies
Bifunctional Molecules Bridging Degradation Pathways
Proteolysis-Targeting Chimeras (PROTACs) represent the most advanced UPS-targeting technology, utilizing bifunctional molecules that simultaneously bind E3 ubiquitin ligases and proteins of interest (POIs), inducing POI ubiquitination and proteasomal degradation [43] [55]. Over 15 PROTAC candidates have entered clinical trials, with ARV-471 (targeting ER for breast cancer) and ARV-110 (targeting AR for prostate cancer) demonstrating promising results in phase II/III trials [55].
Autophagy-Targeting Chimeras (AUTACs) incorporate a degradation tag (e.g., guanine derivatives) that mimics endogenous autophagy signals, recruiting the autophagy machinery to target proteins, protein aggregates, or even organelles [55]. AUTACs demonstrate particular efficacy against pathological aggregates in neurodegenerative disease models.
Autophagosome Tethering Compounds (ATTECs) directly bind both LC3 on autophagosomes and target proteins, effectively "hitching" cargo to forming autophagosomes [55]. This approach has shown promise in degrading lipid droplets and mutant huntingtin protein.
Autophagy-Targeting Chimeras (AUTOTAC) utilize the ZZ domain to bind the p62/SQSTM1 receptor, activating selective autophagy against various POIs, including pathological aggregates and oncoproteins [55].
Lysosome-Targeting Chimeras (LYTACs) extend targeted degradation to extracellular and membrane proteins by conjugating antibody binders to ligands that engage lysosome-shuttling receptors [55].
Nanotechnology-Enhanced Degradation Platforms
Nanomaterial integration addresses key limitations of molecular degraders, including poor solubility, limited bioavailability, and inefficient delivery [43]. Liposomes, polymeric nanoparticles, and inorganic nanoparticles enhance targeted delivery to specific tissues or cellular compartments [43]. Nanozymes generating reactive oxygen species can modulate ubiquitination-deubiquitination balance and enhance endosomal escape, synergizing with degradation technologies [43]. Ferritin-based nanocages enable tissue-specific targeting through intrinsic tropisms [43].
Integrated Degradation Pathways: This diagram illustrates how targeted degradation technologies bridge protein substrates with cellular degradation machinery through molecular chimeras.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for UPS and Autophagy Studies
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | UPS pathway inhibition, substrate stabilization | Reversible (MG132) vs. irreversible (Carfilzomib) inhibition |
| Lysosome Inhibitors | Bafilomycin A1, Chloroquine, NH4Cl | Autophagy flux blockade, cargo accumulation | V-ATPase inhibitor (Bafilomycin) vs. lysosomotropic agents |
| UPS Activity Reporters | Ubiquitin fusion degradation (UFD) substrates, UbG76V-GFP | Real-time monitoring of proteasome function | GFP-based quantitation, flow cytometry compatibility |
| Autophagy Reporters | mRFP-GFP-LC3, GFP-LC3, Cyto-ID | Autophagosome formation and flux measurement | Tandem fluorescent probes distinguish autophagosomes/lysosomes |
| E3 Ligase Modulators | MLN4924 (NAE inhibitor), Nutlin-3 (MDM2 inhibitor) | Specific regulation of ubiquitination pathways | Target specific E3 ligase families or interactions |
| Autophagy Inducers | Rapamycin (mTOR inhibitor), Torin1, Earle's Balanced Salt Solution | Controlled induction of autophagy | mTOR-dependent vs. mTOR-independent mechanisms |
| Ubiquitin Linkage Tools | K48- or K63-specific antibodies, linkage-specific DUBs | Ubiquitin chain topology analysis | Distinguish degradation signals (K48) vs. alternative functions |
| Selective Autophagy Receptors | Recombinant p62, NBR1, OPTN | In vitro reconstitution of selective autophagy | Study receptor-cargo interactions and mechanisms |
The future of targeted protein degradation lies in strategically integrating UPS and autophagy pathways to address their respective limitations while leveraging their complementary strengths. While UPS offers rapid, precise degradation of individual proteins, autophagy provides superior capacity for aggregate clearance and organelle turnover. Emerging technologies that bridge these pathways—such as AUTAC molecules that engage autophagy for cytosolic targets while utilizing UPS principles—demonstrate enhanced efficacy against challenging substrates. The continued development of nano-enabled delivery platforms will further potentiate these approaches by improving pharmacokinetics and tissue-specific targeting. For researchers and drug developers, the strategic selection of degradation pathways should be guided by substrate characteristics (solubility, localization, aggregation propensity), disease context, and desired degradation kinetics. As our understanding of the intricate crosstalk between UPS and autophagy deepens, multi-pathway degradation approaches will increasingly dominate the next generation of therapeutic strategies for proteinopathies, cancer, and other intractable diseases.
Conclusion
The UPS and autophagy are not redundant but complementary pillars of cellular proteostasis, each with distinct strengths. The UPS excels in the rapid, precise degradation of soluble, short-lived proteins, while autophagy is indispensable for clearing bulky cargo like protein aggregates and damaged organelles. The emergence of targeted degradation technologies like PROTACs and AUTACs has transformed these biological pathways into powerful therapeutic platforms. However, challenges remain, including pathway dysregulation in chronic diseases and the need for improved drug-like properties of degrader molecules. Future research must focus on elucidating the detailed molecular crosstalk between these systems, developing strategies to overcome compensatory mechanisms that limit efficacy, and advancing nano-delivery systems for tissue-specific targeting. The intelligent integration of both UPS- and autophagy-based approaches, potentially in a sequential or synergistic manner, holds the greatest promise for treating complex diseases driven by proteostasis failure, such as neurodegenerative disorders and cancer, heralding a new era in precision medicine.
References
- 1. Emerging Paradigm of Crosstalk between Autophagy and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5750708/]
- 2. Crosstalk Between Mammalian Autophagy and the ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2018.00128/full]
- 3. Relationship between the proteasomal system and autophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3627065/]
- 4. Integration of autophagy, proteasomal degradation ... [https://pubmed.ncbi.nlm.nih.gov/23070014/]
- 5. The ubiquitin-proteasome system and autophagy [https://www.sciencedirect.com/science/article/abs/pii/S1357272516301935]
- 6. Cellular Responses to Misfolded Proteins and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4445682/]
- 7. The degradation pathway of a model misfolded protein is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6014095/]
- 8. Inhibition of endoplasmic reticulum associated degradation ... [https://pubmed.ncbi.nlm.nih.gov/23515578/]
- 9. Impact of proteostasis workload on sensitivity to proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12106538/]
- 10. Ubiquitination-Proteasome System (UPS) and Autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8909305/]
- 11. Degradation Mechanism of Autophagy-Related Proteins and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9266641/]
- 12. Lysosome and related protein degradation technologies [https://www.sciencedirect.com/science/article/abs/pii/S1359644623002830]
- 13. p62/SQSTM1 and Selective Autophagy in Cardiometabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6653798/]
- 14. Orchestration of selective autophagy by cargo receptors [https://www.sciencedirect.com/science/article/pii/S0960982222017584]
- 15. Autophagy–Lysosome Pathway in Multiple System Atrophy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562437/]
- 16. Cargo recognition and degradation by selective autophagy [https://www.nature.com/articles/s41556-023-01177-x]
- 17. Autophagy Related Protein 5 - an overview [https://www.sciencedirect.com/topics/neuroscience/autophagy-related-protein-5]
- 18. Direct Experimental Evidence for Selective Uptake of Function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12538810/]
- 19. of Degradation in neurodegenerative diseases... misfolded proteins [https://www.nature.com/articles/emm2014117?error=cookies_not_supported]
- 20. The Roles of Ubiquitin in Mediating Autophagy [https://www.mdpi.com/2073-4409/9/9/2025]
- 21. Targeted protein and protein-condensate degradation in ... [https://www.sciencedirect.com/science/article/abs/pii/S1674205225002060]
- 22. Discovery of Evolutionary Loss of the Ubiquitin-like ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11764061/]
- 23. The Role of WIPI2, ATG16L1 and ATG12-ATG5 in ... [https://www.sciencedirect.com/science/article/pii/S0022283625002049]
- 24. Reduced UPF1 levels in senescence impair nonsense- ... [https://www.nature.com/articles/s42003-025-07502-4]
- 25. Exploring the Molecular Mechanisms of Autophagy-related ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12208648/]
- 26. Small molecule targeted protein via the degradation : venturing... UPS [https://pubs.rsc.org/en/content/articlelanding/2025/md/d4md00718b]
- 27. Crosstalk Between Mammalian Autophagy and the Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6175981/]
- 28. Ubiquitination and selective autophagy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3524631/]
- 29. Atg8 family proteins, LIR/AIM motifs and other interaction modes [https://pmc.ncbi.nlm.nih.gov/articles/PMC7615515/]
- 30. The Roles of Ubiquitin in Mediating Autophagy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7564124/]
- 31. Mechanistic insights into selective autophagy pathways [https://pmc.ncbi.nlm.nih.gov/articles/PMC5549613/]
- 32. -mediated regulation of Ubiquitin | Journal of Biomedical... autophagy [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-019-0569-y]
- 33. The Ubiquitin Ligase Smurf1 Functions in Selective of... Autophagy [https://pubmed.ncbi.nlm.nih.gov/28017659/]
- 34. VPS34 K29/ K branched 48 ... | Nature Communications ubiquitination [https://www.nature.com/articles/s41467-021-21715-1?error=cookies_not_supported&code=e00e3cf4-ed4c-42c1-9604-db593a955e35]
- 35. Ubiquitination and selective autophagy | Cell Death & ... [https://www.nature.com/articles/cdd201272]
- 36. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 37. Endoplasmic reticulum stress: molecular mechanism and ... [https://www.nature.com/articles/s41392-023-01570-w]
- 38. The interconnective role of the UPS and autophagy in ... [https://pubmed.ncbi.nlm.nih.gov/39800773/]
- 39. Autophagy hub-protein p62 orchestrates oxidative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12167074/]
- 40. Proteostasis control by the unfolded protein response - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5546321/]
- 41. A Systematic Protein Turnover Map for Decoding Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7745259/]
- 42. The N-degron pathway governs autophagy to promote ... [https://www.nature.com/articles/s41467-025-61191-5]
- 43. Targeted protein degradation technologies: Emerging ... [https://www.sciopen.com/article/10.26599/NR.2025.94907983]
- 44. Targeted Protein Degradation with PROTACs and ... [https://blog.crownbio.com/targeted-protein-degradation-with-protacs-and-molecular-glues]
- 45. Review Molecular glues and PROTACs in targeted protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225005623]
- 46. Targeting the Ubiquitin–Proteasome System for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495026/]
- 47. Molecular Glues: The Adhesive Connecting Targeted Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9910052/]
- 48. PROTAC 2.0: Expanding the frontiers of targeted protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000893]
- 49. PROTAC-induced protein structural dynamics in targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11867615/]
- 50. Inside the Humanized CRBN Mouse Model [https://biocytogen.com/blogs/crbn-humanized-mice-enable-molecular-glue-and-protac-research]
- 51. A degron-mimicking molecular glue drives CRBN homo- ... [https://www.nature.com/articles/s41467-025-65094-3]
- 52. Molecular glue-mediated targeted protein degradation [https://www.sciencedirect.com/science/article/pii/S2589004224019370]
- 53. The Autophagy-Lysosomal Pathways and Their Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6356230/]
- 54. Targeted protein degradation via the autophagy-lysosome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9397417/]
- 55. Targeted Degradation Technologies Utilizing Autophagy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295361/]
- 56. Challenges and opportunities for lysosome-targeting ... [https://www.sciencedirect.com/science/article/pii/S2666386425003777]
- 57. Targeted protein degradation directly engaging lysosomes or ... [https://pubs.rsc.org/en/content/articlehtml/2024/cs/d3cs00344b]
- 58. Emerging Concepts of Targeted Protein Degrader ... [https://www.mdpi.com/1422-0067/26/12/5582]
- 59. Rational design of the linkers in targeting chimeras - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451458/]
- 60. The AUTOTAC chemical biology platform for targeted ... [https://www.nature.com/articles/s41467-022-28520-4]
- 61. 10-evolved-terms-every-scientist-should-know-about ... [https://resources.nanotempertech.com/blog/10-evolved-terms-every-scientist-should-know-about-targeted-protein-degradation-in-2025]
- 62. Chaperone-mediated autophagy: Molecular mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2914824/]
- 63. Chaperone‐mediated autophagy: Molecular mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10466100/]
- 64. Chaperone-mediated autophagy manipulates PGC1α ... [https://www.nature.com/articles/s41467-025-59618-0]
- 65. Chaperone-mediated autophagy directs a dual mechanism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162876/]
- 66. The dual role of chaperone-mediated autophagy in ... [https://www.sciencedirect.com/science/article/pii/S1040842825000885]
- 67. Targeted protein degradation: advances in drug discovery ... [https://www.nature.com/articles/s41392-024-02004-x]
- 68. Autophagy mediated targeting degradation, a promising ... [https://www.sciencedirect.com/science/article/abs/pii/S0045206824003717]
- 69. Nanoparticle‐Mediated Targeted Protein Degradation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12232892/]
- 70. p62 links the autophagy pathway and the ubiqutin ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-016-0031-z]
- 71. Evaluating cell cycle- and autophagy-associated cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12216923/]
- 72. Review article CNS delivery of targeted protein degraders [https://www.sciencedirect.com/science/article/pii/S0168365924004176]
- 73. Degradation of misfolded proteins by ubiquitin-proteasome ... [https://www.sciencedirect.com/science/article/abs/pii/B9780443187162000077]
- 74. Interplay between the ubiquitin-proteasome system and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2856956/]
- 75. Dynamics of the Degradation of Ubiquitinated Proteins by ... [https://www.sciencedirect.com/science/article/pii/S0021925819489539]
- 76. How Does the Battle between the UPS and Autophagy ... [https://www.mdpi.com/1422-0067/24/3/2221]
- 77. Proteasome Inhibition Activates Autophagy-Lysosome ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2019.00170/full]
- 78. Review Autophagy and the ubiquitin-proteasome system [https://www.sciencedirect.com/science/article/pii/S0925443908001944]
- 79. Role of TRIM24 in the regulation of proteasome-autophagy ... [https://www.nature.com/articles/s41420-025-02355-6]
- 80. Proteostasis Disruption by Proteasome Inhibitor MG132 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12256276/]
- 81. Modulation of the Ubiquitin-Proteasome System by ... [https://www.sciencedirect.com/science/article/pii/S095528632500289X]
- 82. Integrated transcriptomic analysis identifies lysosomal ... [https://www.nature.com/articles/s41598-025-20595-5]
- 83. Molecular switching from ubiquitin-proteasome to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6928553/]
- 84. Autophagy: The convergence point of aging and cancer [https://www.sciencedirect.com/science/article/pii/S2405580825000731]
- 85. A bioinformatic investigation of proteasome and autophagy ... [https://www.sciencedirect.com/science/article/pii/S2405844023053963]
- 86. The role of autophagy in fibrosis: Mechanisms, progression ... [https://www.spandidos-publications.com/10.3892/ijmm.2025.5502]
- 87. Transmission of unfolded protein response—a regulator ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11796368/]
- 88. The Unfolded Protein Response—Novel Mechanisms ... [https://www.mdpi.com/2072-6694/17/22/3639]
- 89. Degradation of misfolded proteins in neurodegenerative ... [https://www.nature.com/articles/emm2014117]
- 90. A Method for the Acute and Rapid Degradation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5733393/]
- 91. Rapid and reversible knockdown of endogenous proteins by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3937121/]
- 92. Targeted Protein Degradation: Advances, Challenges, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10498452/]
- 93. From PROTAC to TPD: Advances and Opportunities in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10818447/]
- 94. Novel protein complexes containing autophagy and UPS ... [https://www.nature.com/articles/s41419-022-05339-x]
- 95. Protein quality control systems in neurodegeneration [https://pmc.ncbi.nlm.nih.gov/articles/PMC12441361/]
- 96. Autophagy and the ubiquitin-proteasome system [https://pmc.ncbi.nlm.nih.gov/articles/PMC2621359/]
- 97. Protein Misfolding and Aggregation in Proteinopathies [https://pmc.ncbi.nlm.nih.gov/articles/PMC9944956/]
- 98. Autophagic Clearance of PolyQ Proteins Mediated by ... [https://www.sciencedirect.com/science/article/pii/S0092867414008083]
- 99. Novel protein complexes containing autophagy and UPS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9649694/]
- 100. Selective Autophagy of Macromolecular Complexes [https://www.sciencedirect.com/science/article/pii/S0022283624001694]
- 101. Methods for measuring misfolded protein clearance in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6988565/]
- 102. Analysis of protein and lipid dynamics using confocal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3538152/]
- 103. Nuclear proteasomes as a backup for autophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC11702925/]
- 104. Interlinked destinies: How ubiquitin-proteasome and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11283956/]
- 105. Developing Effective Degrader Compounds: Why Cellular ... [https://www.promega.kr/resources/pubhub/2025/developing-effective-degrader-compounds-why-cellular-degradation-kinetics-are-key/]
- 106. Interplay between unfolded protein response and autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4579870/]
- 107. Autophagy and the unfolded protein response shape ... [https://www.sciencedirect.com/science/article/abs/pii/S0026049524000374]
- 108. Contemporary trends in targeted protein degradation for ... [https://pubmed.ncbi.nlm.nih.gov/40914014/]
- 109. Molecular Design of Novel Protein-Degrading ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12196083/]
- 110. Development of a targeted BioPROTAC degrader selective ... [https://www.nature.com/articles/s41467-025-65481-w]
- 111. New class of protein misfolding simulated in high definition [https://science.psu.edu/news/Obrien8-2025]
- 112. Autophagy Counterbalances Endoplasmic Reticulum ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0040423]
- 113. Novel protein complexes containing autophagy and UPS components... [https://www.nature.com/articles/s41419-022-05339-x?error=cookies_not_supported]
- 114. Autophagy as a potential therapeutic target in regulating ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1579183/full]
- 115. Mechanistic insights and therapeutic strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12018664/]
- 116. Crosstalk between acetylation modification and autophagy ... [https://www.nature.com/articles/s41420-025-02809-x]
- 117. Crosstalk and Interplay between the Ubiquitin-Proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5547213/]
- 118. Targeted protein and protein-condensate degradation in ... [https://www.sciencedirect.com/science/article/pii/S1674205225002060]
- 119. Ubiquitination and Proteasomal Degradation of ATG12 ... [https://pubmed.ncbi.nlm.nih.gov/25629932/]
- 120. A hidden talent for a RNA decay factor: UPF1 directs protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5843485/]