Unlocking Complex Biologics: Strategies for Robust Disulfide Bond Formation in E. coli Cytoplasm
This article provides a comprehensive guide for researchers and biopharmaceutical developers aiming to produce disulfide-rich therapeutic proteins in E.
Unlocking Complex Biologics: Strategies for Robust Disulfide Bond Formation in E. coli Cytoplasm
Abstract
This article provides a comprehensive guide for researchers and biopharmaceutical developers aiming to produce disulfide-rich therapeutic proteins in E. coli cytoplasm. We first explore the fundamental challenge of the reducing cytoplasmic environment. We then detail cutting-edge methodologies, including engineered strains and redox pathway manipulation, for enabling correct disulfide bond formation. The guide covers common troubleshooting scenarios and optimization techniques for yield and fidelity. Finally, it presents validation frameworks and comparative analyses of leading systems (e.g., SHuffle, CyDisCo) to inform strategic choices. The synthesis offers a roadmap for advancing the cytoplasmic production of antibodies, cytokines, and other complex biologics.
The Disulfide Dilemma: Why the E. coli Cytoplasm is Reducing and Why It Matters for Protein Therapeutics
The Critical Role of Disulfide Bonds in Protein Stability, Function, and Therapeutics
Troubleshooting Guide & FAQs for Enhancing Disulfide Bond Formation inE. coliCytoplasm
FAQ 1: My target protein expressed in the E. coli cytoplasm is completely insoluble. What are the first steps I should take? Answer: Cytoplasmic insolubility often indicates improper folding due to an overly reducing environment preventing disulfide bond formation. First, verify the protein's sequence for an even number of cysteines. Then, switch to an engineered E. coli strain designed for cytoplasmic disulfide bond formation, such as SHuffle T7 or Origami 2. Ensure you are using a low-temperature induction protocol (e.g., 16-25°C post-IPTG addition) to slow translation and allow folding.
FAQ 2: I am using an SHuffle strain, but my protein yield is very low. How can I improve it? Answer: Low yield in disulfide-competent strains can result from metabolic burden or residual toxicity. Consider the following adjustments:
- Induction Optimization: Reduce IPTG concentration (e.g., 0.1-0.5 mM) and induce at lower cell density (OD600 ~0.4-0.6).
- Media: Use rich media like TB or 2xYT supplemented with 0.5% glucose to repress expression until induction.
- Promoter: If using a T7 system, ensure the strain carries the pLysS plasmid for tighter repression, reducing leaky expression toxicity.
FAQ 3: How do I definitively confirm that the correct intramolecular disulfide bonds have formed in my purified protein? Answer: Use a combination of analytical techniques:
- Non-Reducucing vs. Reducing SDS-PAGE: A faster migration under non-reducing conditions suggests a more compact, disulfide-bonded structure.
- Mass Spectrometry (Intact Protein): The observed mass under non-reducing conditions should match the theoretical mass minus 2 Da per disulfide bond (loss of H2).
- Peptide Mapping LC-MS/MS: Digest the protein with an enzyme like trypsin under non-reducing conditions, then analyze by LC-MS/MS. Cysteines linked by a disulfide bond will appear as a single peptide with a combined mass, confirming the specific bond pairing.
FAQ 4: My protein forms aggregates or incorrect intermolecular disulfide bonds. How can I promote correct intramolecular bonding? Answer: Incorrect intermolecular bonding (aggregation) suggests cysteine thiols are oxidizing randomly. To guide correct pairing:
- Redox Tuning: Supplement your growth medium with redox agents. A low ratio of reduced to oxidized glutathione (e.g., GSH:GSSG at 5:1 to 1:1) can provide a oxidizing "shuffle" to correct mispaired bonds.
- Molecular Chaperone Co-expression: Co-express foldase chaperones like DsbC (which also has isomerase activity) in the cytoplasm using compatible plasmids. This is crucial for proteins with multiple disulfides.
- Sequential Secretion Strategy: If applicable, consider expressing your protein with a signal sequence to direct it to the E. coli periplasm, the natural compartment for disulfide bond formation.
Key Experimental Protocol: Analyzing Disulfide Bond Formation via Non-Reducing/Reducing SDS-PAGE
Objective: To quickly assess the oxidation state and oligomerization status of a recombinant protein expressed in the E. coli cytoplasm.
Materials:
- Cell pellet from induced culture.
- Lysis Buffer: 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 100 µg/mL lysozyme, optional: protease inhibitors.
- 2X Non-Reducing Sample Buffer: 125 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 0.01% Bromophenol Blue.
- 2X Reducing Sample Buffer: As above, plus 10% β-mercaptoethanol (added fresh) or 100 mM DTT.
- Heating block or water bath.
- SDS-PAGE gel (appropriate % acrylamide for your protein).
Procedure:
- Lysis: Resuspend cell pellet in Lysis Buffer. Incubate on ice for 30 minutes. Clarify by centrifugation (13,000 x g, 20 min, 4°C). Transfer supernatant (soluble fraction) to a new tube.
- Sample Preparation:
- Prepare two tubes for each sample: "Non-Reduced" (NR) and "Reduced" (R).
- Mix equal volumes (e.g., 20 µL) of protein sample with 2X Non-Reducing Buffer (NR tube) and with 2X Reducing Buffer (R tube).
- Denaturation: Heat the Reduced sample at 95-100°C for 5-10 minutes. Heat the Non-Reduced sample at 70°C for 10 minutes only. (Avoid boiling non-reduced samples with SDS, as it can promote artificial disulfide scrambling).
- Electrophoresis: Load equal volumes of NR and R samples on adjacent lanes of an SDS-PAGE gel. Run the gel at constant voltage.
- Analysis: Compare migration patterns. A protein with intramolecular disulfides will migrate faster in the NR lane than in the R lane. A smear or high molecular weight band in the NR lane suggests aggregation via intermolecular disulfides.
Research Reagent Solutions Toolkit
| Reagent / Strain | Primary Function in Cytoplasmic Disulfide Bond Research |
|---|---|
| SHuffle T7 Express E. coli | Engineered strain with a trxB/gor double mutation (oxidizing cytoplasm) and chromosomal DsbC expression for isomerization. Ideal for cytoplasmic expression. |
| Origami 2 E. coli | trxB/gor double mutant strain, providing an oxidizing cytoplasm. Often used with a periplasmic targeting vector but can be used for cytoplasmic work. |
| pBAD-DsbC Plasmid | Plasmid for arabinose-inducible expression of DsbC chaperone/isomerase. Can be co-transformed to enhance correct folding in the cytoplasm. |
| Reduced (GSH) & Oxidized (GSSG) Glutathione | Used to fine-tune the redox potential of the growth medium or lysis buffer to promote oxidation or isomerization of disulfides. |
| β-Mercaptoethanol (BME) / Dithiothreitol (DTT) | Strong reducing agents used in sample buffers to break all disulfide bonds for comparative analysis. |
| Iodoacetamide (IAM) | Alkylating agent used to permanently block free cysteine thiols, preventing artificial disulfide scrambling during sample preparation. |
| Cysteine-Cysteine Disulfide Bond (Standard) | HPLC standard used for calibrating analytical methods to quantify disulfide bond content or stability. |
Table 1: Comparison of Common E. coli Strains for Recombinant Disulfide Bond Formation
| Strain | Genotype | Key Feature | Best Application | Typical Yield Impact |
|---|---|---|---|---|
| BL21(DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS | Standard, reducing cytoplasm | Proteins with no/few disulfides | Baseline (High) |
| Origami 2 | ∆(ara-leu) ∆lacX74 ∆phoA PvuII phoR araD139 ahpC galE galK rpsL F'[lac+ lacIq pro] (DE3) gor522::Tn10 trxB | trxB/gor double mutant; oxidizing cytoplasm | Periplasmic expression or less complex cytoplasmic disulfides | Moderate Reduction |
| SHuffle T7 Express | ∆trxB ∆gor ∆ahpC sulA::kanR, chromosomal DsbC | Oxidizing cytoplasm + cytosolic DsbC isomerase | Complex, multi-disulfide proteins in the cytoplasm | Variable, can be low |
Table 2: Effect of Redox Buffer Supplementation on Soluble Yield of a Model Protein (scFv Antibody Fragment) in SHuffle Strain
| Supplement in TB Media (at induction) | GSH:GSSG Ratio (Approx.) | Final Soluble Yield (mg/L) | Correctly Folded (by ELISA) |
|---|---|---|---|
| None (Control) | N/A | 2.1 | 15% |
| 5 mM GSH | All Reduced | 1.8 | 10% |
| 1 mM GSSG | All Oxidized | 3.5 | 40% |
| 2.5 mM GSH + 0.5 mM GSSG | 5:1 | 5.2 | 75% |
| 1 mM GSH + 1 mM GSSG | 1:1 | 4.1 | 85% |
Visualizations
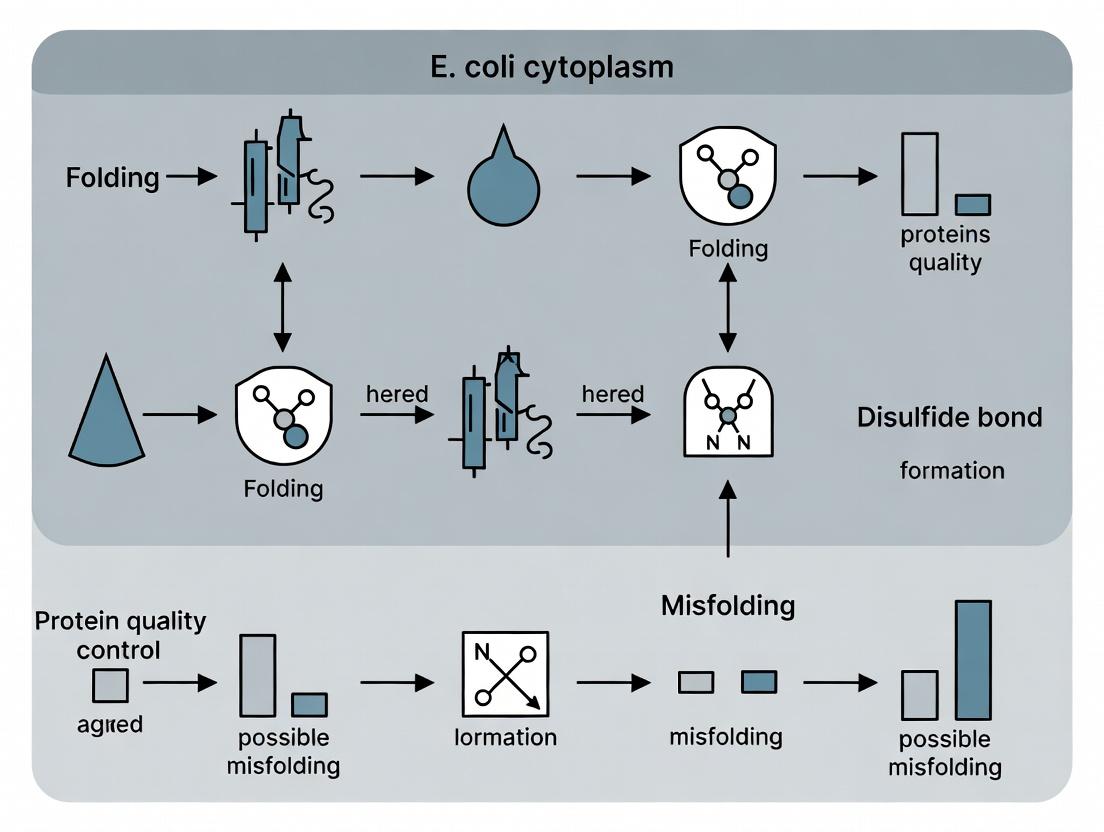
Title: Workflow for Cytoplasmic Disulfide Bond Expression & Analysis
Title: Disulfide Bond Folding Pathways in Engineered E. coli
Technical Support Center: Troubleshooting Disulfide Bond Formation in the E. coli Cytoplasm
FAQs & Troubleshooting Guides
Q1: My protein of interest shows low yield and aggregation when expressed in the standard E. coli cytoplasm. What is the primary redox issue? A: The standard E. coli cytoplasm is a reducing environment maintained by the thioredoxin (Trx) and glutaredoxin (Grx) pathways. These systems actively reduce incorrectly formed disulfide bonds, preventing proper folding of proteins that require stable, structural disulfides. Your target protein is likely being misfolded due to premature reduction.
Q2: I have knocked out the trxB and gor genes to disrupt the reductive pathways, but my protein still isn't forming disulfide bonds efficiently. What else should I check?
A: Double-check your strain genotype. Ensure both trxB (thioredoxin reductase) and gor (glutathione reductase) are completely inactivated. Residual activity can compromise the oxidative environment. Additionally, consider the following:
- Oxidative Damage: The
ΔtrxB Δgorstrain grows very slowly due to hypersensitivity to oxidative stress. This can reduce cell viability and protein yield. - Lack of an Oxidizing Catalyst: The cytoplasm may lack sufficient oxidative power to form disulfides rapidly. Co-express a sulfhydryl oxidase (e.g., Erv1p) or an engineered disulfide bond isomerase (DsbC) in the cytoplasm.
- Incorrect Disulfide Pairing: Without proper isomerization, non-native disulfides can form, leading to aggregation.
Q3: What is the quantitative difference in redox potential between the standard cytoplasm and the periplasm? A: The redox potential is a quantitative measure of the reducing/oxidizing power of a compartment. See the table below for a comparison.
Table 1: Redox Potential of E. coli Compartments
| Compartment | Typical Redox Potential (mV) | Dominant Redox System | Suitability for Disulfide Bond Formation |
|---|---|---|---|
| Cytoplasm (Wild-type) | -270 to -290 mV | Thioredoxin & Glutaredoxin (Reduced) | Poor - Strongly Reducing |
| Cytoplasm (ΔtrxB Δgor) | -205 to -230 mV | Oxidized Glutathione (GSSG) Accumulates | Moderate - Weakly Reducing |
| Periplasm | -165 to -185 mV | DsbA/DsbB System (Oxidized) | Good - Oxidizing |
Q4: How do I measure the effect of my genetic modifications on the intracellular redox state? A: Use a redox-sensitive GFP (roGFP) biosensor. roGFP exhibits a shift in fluorescence excitation ratio (400 nm / 480 nm) upon oxidation/reduction, allowing in vivo measurement of redox potential.
Experimental Protocol: Assessing Cytoplasmic Redox State with roGFP2
- Clone: Fuse roGFP2 to your expression plasmid or a compatible reporter plasmid.
- Transform: Introduce the roGFP2 plasmid into your test strains (e.g., wild-type,
ΔtrxB Δgor,ΔtrxB Δgorwith oxidase expression). - Culture & Measure: Grow cultures to mid-log phase (OD600 ~0.6). Harvest cells and resuspend in PBS.
- Fluorescence Reading: Load samples into a fluorescence microplate reader. Measure fluorescence intensity with excitation at 400 nm and 480 nm, and emission at 510 nm.
- Calculate Ratio: Compute the ratio F400/F480 for each sample. A higher ratio indicates a more oxidized environment. Calibrate with fully reduced (DTT-treated) and fully oxidized (H2O2-treated) cell samples to determine the relative redox state.
Q5: Glutathione is central to the Grx system. How do its levels change in engineered strains, and how can I monitor this?
A: Disrupting the gor gene blocks the reduction of oxidized glutathione (GSSG), leading to a buildup of GSSG and a decrease in the reduced glutathione (GSH) pool. This alters the GSH:GSSG ratio, a key redox buffer. Monitor this using commercial glutathione assay kits (e.g., colorimetric DTNB-based assays).
Table 2: Key Redox Metabolite Changes in Engineered E. coli Strains
| Strain | GSH Level | GSSG Level | GSH:GSSG Ratio (Approx.) | Cytoplasmic Redox State |
|---|---|---|---|---|
| Wild-type (e.g., BL21) | High (~10 mM) | Very Low | >200:1 | Strongly Reducing |
ΔtrxB only |
Moderately High | Low | ~50:1 | Reducing |
Δgor only |
Low | High | ~3:1 | Oxidized (Stressed) |
ΔtrxB Δgor (e.g., SHuffle) |
Low | Very High | <1:10 | Oxidizing (Conducive for disulfides) |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Cytoplasmic Disulfide Bond Research
| Item | Function/Description | Example/Catalog Consideration |
|---|---|---|
| E. coli Strain: SHuffle T7 | A ΔtrxB Δgor strain with a periplasmic DsbC expressed in the cytoplasm to catalyze disulfide bond formation and isomerization. | New England Biolabs, C3029J |
| Redox-Sensitive GFP (roGFP2) | A genetically encoded biosensor for real-time, non-invasive measurement of cellular redox potential. | Addgene, plasmid #64976 |
| Glutathione Assay Kit (DTNB) | Quantifies total, reduced (GSH), and oxidized (GSSG) glutathione levels in cell lysates. | Sigma-Aldrich, CS0260 |
| Insoluble Protein Extraction Reagent | For solubilizing aggregated protein from inclusion bodies for analysis (e.g., urea, guanidine HCl). | Thermo Fisher Scientific, 78501 |
| Anti-DsbA / Anti-DsbC Antibodies | Western blotting to verify the expression and localization of key redox pathway proteins. | Lab-made or commercial (e.g., Abcam) |
| IPTG | Inducer for T7/lac-based expression systems to control the timing of target protein expression. | Gold Biotechnology, I2481C |
| 2-Mercaptoethanol (BME) / DTT | Reducing agents for control experiments to break disulfide bonds in SDS-PAGE sample prep. | Sigma-Aldrich, M6250 / D0632 |
| Copper Phenanthroline (CuPhe) | A membrane-permeable chemical oxidant used to artificially induce disulfide formation in the cytoplasm. | Sigma-Aldrich, 146625 |
Pathway & Workflow Diagrams
Troubleshooting Guide & FAQs
FAQ 1: My recombinant protein shows no disulfide bonds despite using a trxB-/gor- strain. What could be wrong?
Answer: The double knockout of thioredoxin reductase (trxB) and glutathione reductase (gor) is foundational but often insufficient alone. Common issues include:
- Residual Reductive Pathways: The glutaredoxin system may still be active, scavenging oxidizing equivalents. Consider adding a mutation in
grxA/goror using a strain with an additionalahpC*mutation (e.g., SHuffle strains) to further oxidize the cytoplasm. - Protein Aggregation: Misfolded, aggregation-prone intermediates can form. Check solubility and co-express chaperones like
DsbC(which also has isomerase activity) in the cytoplasm using a signal sequence knockout variant (e.g.,DsbCΔSS). - Incorrect Disulfide Pairing: The cytoplasm may lack sufficient isomerase activity. Co-express a catalyst like eukaryotic Protein Disulfide Isomerase (PDI) or bacterial
DsbCΔSSto shuffle incorrect bonds.
FAQ 2: How do I quantify the oxidative state of the cytoplasm in my engineered strain?
Answer: Use a redox-sensitive green fluorescent protein (roGFP). roGFP2 is a genetically encoded biosensor that reports on the glutathione redox potential (EGSH). The ratio of fluorescence after excitation at 400 nm and 480 nm is calibrated to the redox state.
Protocol: Quantifying Cytoplasmic Redox Potential with roGFP2
- Clone & Express: Fuse roGFP2 to your protein or express it freely in the cytoplasm of your engineered E. coli strain.
- Culture & Harvest: Grow cells to mid-log phase (OD600 ~0.6) under your experimental conditions. Harvest 1 mL aliquots by centrifugation.
- Measure Fluorescence: Resuspend cells in PBS. Immediately measure fluorescence intensity in a plate reader or spectrophotometer with excitation at 400 nm and 480 nm, and emission at 510 nm.
- Calculate Ratio: For each sample, calculate the ratio R = Fluorescence(400nm ex)/Fluorescence(480nm ex).
- Normalize: To determine the degree of oxidation (OxD), treat parallel samples with 10 mM DTT (fully reduced) and 10 mM Diamide (fully oxidized). Apply the formula:
OxD = (R - R_reduced) / (R_oxidized - R_reduced) - Interpret: An OxD value closer to 1 indicates a more oxidized cytoplasm.
Table 1: Redox Potential Indicators with roGFP2
| Strain Background | Typical Application | Approximate Cytoplasmic EGSH (mV) | roGFP2 OxD Range |
|---|---|---|---|
| Wild-type (e.g., BL21) | Baseline reducing environment | -270 to -300 | 0.1 - 0.3 |
| trxB- gor- (e.g., Origami) | Enhanced disulfide bond formation | -220 to -250 | 0.4 - 0.7 |
| trxB- gor- ahpC* (e.g., SHuffle) | Highly oxidative cytoplasm for complex proteins | -180 to -210 | 0.7 - 0.9 |
FAQ 3: What are the best practices for expressing a eukaryotic protein with multiple disulfides in the E. coli cytoplasm?
Answer: A systematic approach combining strain selection, fusion tags, and co-expression factors is required.
- Strain Selection: Start with a strain engineered for cytoplasmic oxidation (e.g.,
trxB-gor-ahpC*dsbC). - Use a Solubility-Enhancing Fusion Tag: Fuse your target protein to tags like E. coli TrxA (thioredoxin) or SUMO. These tags improve solubility and can themselves be engineered to influence the local redox environment.
- Co-express Chaperones and Isomerases: Co-express cytoplasmic variants of disulfide isomerases (e.g.,
DsbCΔSS,PDI) and chaperones (e.g.,GroEL/GroES,DnaK/DnaJ/GrpE) to aid folding and correct pairing. - Optimize Expression Conditions: Use lower growth temperatures (25-30°C), induce at lower cell densities (OD600 ~0.4-0.6), and consider auto-induction media to slow protein production and favor folding.
Table 2: Key Research Reagent Solutions
| Reagent / Strain | Primary Function | Example Use Case |
|---|---|---|
| SHuffle T7 Express (C3029J) | Combines trxB-, gor-, ahpC*, and cytoplasmic DsbC. Provides oxidative folding and isomerase activity. |
Expression of eukaryotic proteins with multiple/complex disulfides. |
| pET-39b(+) Vector (Novagen) | Expresses target protein as a DsbA fusion, directing it to the oxidizing periplasm. | Periplasmic expression of disulfide-bonded proteins. |
| pBAD Vector System (Invitrogen) | Allows tight, tunable expression with arabinose. Critical for expressing toxic proteins. | Fine-control over expression of redox enzymes or toxic targets. |
| CyDisCo System (Plasmid Set) | Co-expression of eukaryotic PDI and Erv1p sulfhydryl oxidase in the cytoplasm. | High-yield cytoplasmic production of human cytokines & antibodies. |
| roGFP2 Plasmids (Addgene #49435, etc.) | Genetically encoded biosensor for real-time measurement of cellular glutathione redox potential. | Quantifying the effectiveness of cytoplasm oxidation strategies. |
| His-SUMO Fusion Tags | Enhances solubility and offers a cleavage site via Ulp1 protease for tag removal. | Improving yield and solubility of aggregation-prone targets. |
FAQ 4: My protein is expressed but inactive. How do I diagnose misfolded disulfides?
Answer: Inactivity often stems from non-native disulfide bonds. Follow this diagnostic workflow:
- Check Solubility: Perform a simple lysis and centrifugation. If the protein is in the pellet, misfolding/aggregation is likely.
- Analyze Mobility: Run non-reducing vs. reducing SDS-PAGE. A faster mobility under non-reducing conditions typically indicates the presence of some disulfides (more compact structure). A smear or multiple bands suggest heterogeneity in disulfide bonding.
- Perform a Redox Western Blot: Use a differential cysteine alkylation assay with agents like AMS (4-acetamido-4'-maleimidylstilbene-2,2'-disulfonic acid), which increases molecular weight proportionally to the number of free thiols.
- Confirm with Mass Spectrometry: For definitive identification, use LC-MS/MS after protease digestion (e.g., trypsin) under non-reducing conditions to map the specific cysteine linkages.
Diagram Title: Diagnostic Workflow for Suspected Disulfide Misfolding
Diagram Title: Key Pathways in Engineered E. coli Redox System
Technical Support Center: Troubleshooting & FAQs
This support center addresses common experimental challenges in the context of enhancing cytoplasmic disulfide bond formation in E. coli for therapeutic protein production.
Frequently Asked Questions
Q1: My target protein with multiple disulfide bonds is completely insoluble when expressed in the standard cytosol (e.g., in BL21(DE3)). What are my first steps?
A: This is the primary motivation for moving to engineered cytoplasmic systems. First, verify the expression system. Switch to an E. coli strain engineered for cytoplasmic disulfide bond formation, such as SHuffle T7 Express or Origami B(DE3). These strains have a mutated thioredoxin reductase (trxB) and/or glutathione reductase (gor) background, creating a more oxidizing cytoplasm, and often express a disulfide bond isomerase (DsbC) in the cytosol. Always co-express your target with these strains; using a standard strain will lead to aggregation.
Q2: I've switched to an SHuffle strain, but my yield is still low. What should I optimize?
A: Focus on expression conditions. Key parameters to test are:
- Induction Temperature: Lower the temperature post-induction to 16-25°C. Slower protein synthesis favors correct folding.
- Induction Timing: Induce at a lower OD600 (0.4-0.6) to reduce metabolic burden.
- Inducer Concentration: Titrate IPTG (e.g., 0.01-0.5 mM) to find the minimum effective dose.
- Media: Test rich media (e.g., TB) versus minimal media. Rich media often supports higher biomass but can increase acidity; monitor and control pH at 7.0.
Q3: I am seeing proteolytic degradation of my soluble, disulfide-bonded protein in the cytoplasm. How can I mitigate this?
A: The oxidizing cytoplasm can sometimes expose degradation motifs. Use protease-deficient strain derivatives (e.g., SHuffle T7 Express lon ompT). Include protease inhibitor cocktails in your lysis buffer (specific for E. coli proteases). Purify immediately after cell harvest or flash-freeze cell pellets at -80°C. Increasing the expression rate slightly (via slightly higher IPTG) can sometimes outpace degradation, but balance this against aggregation risks.
Q4: How do I definitively confirm that my cytoplasmic protein has formed the correct disulfide bonds?
A: Use a combination of analytical techniques:
- Non-Reducucing vs. Reducing SDS-PAGE: A faster migration under non-reducing conditions suggests intramolecular disulfide formation.
- Mass Spectrometry (Intact Mass): The observed mass under non-reducing conditions should match the theoretical mass with all disulfides, confirming the absence of free cysteines.
- Peptide Mapping with LC-MS/MS: After enzymatic digestion under non-reducing conditions, this identifies which specific cysteines are linked.
Q5: For large-scale fermentation, what are the critical process parameters when using these engineered cytoplasmic strains?
A: Scalability is a key advantage over periplasmic production. Critical parameters include:
- Oxygen Transfer Rate (OTR): Crucial for cell growth and redox metabolism. Ensure adequate agitation and aeration.
- pH Control: Maintain at 7.0 rigorously; use ammonia gas or hydroxide solutions for base.
- Substrate Feed Rate (in fed-batch): Controlled, exponential feeding prevents acetate accumulation, which is more detrimental to these metabolically compromised (trxB/gor) strains.
- Dissolved Oxygen (DO) Spikes: Can indicate metabolic shifts; a common strategy is to let DO rise slightly post-induction to reduce metabolic stress.
Troubleshooting Guide: Common Issues and Solutions
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Low Solubility | Expression in non-engineered strain; Too fast protein synthesis; Lack of chaperones. | 1. Use SHuffle/Origami strains. 2. Reduce induction temperature to 16-25°C. 3. Co-express chaperones (e.g., GroEL/ES, DnaK/J). |
| Low Yield | Proteolytic degradation; Poor cell growth; Inefficient lysis. | 1. Use protease-deficient strain variant. 2. Optimize media (use TB). 3. Use lysozyme + mechanical lysis (sonication, French press). |
| Incorrect Disulfides | Cytoplasm lacks isomerase activity; Oxidation too rapid. | 1. Ensure strain expresses cytoplasmic DsbC (e.g., SHuffle). 2. Add redox mediators (e.g., GSH/GSSG mix) to culture. |
| Poor Scalability | Acetate accumulation; Oxidative stress in bioreactor. | 1. Implement controlled fed-batch with limiting carbon feed. 2. Monitor DO and pH closely; avoid microaerobic zones. |
Experimental Protocol: Expression & Analysis of a Disulfide-Bonded Protein in EngineeredE. coliCytoplasm
Objective: Express, solubly produce, and verify the disulfide bond formation of a target protein in the E. coli cytoplasm.
Materials:
- Strain: E. coli SHuffle T7 Express (NEB #C3029J).
- Vector: pET-based plasmid with target gene.
- Media: LB or Terrific Broth (TB) with appropriate antibiotic (e.g., 100 µg/mL ampicillin).
- Inducer: 0.1-1.0 M Isopropyl β-d-1-thiogalactopyranoside (IPTG) stock.
- Lysis Buffer: 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mg/mL lysozyme, 1x protease inhibitor cocktail, 1 mM PMSF. Do not add DTT or β-mercaptoethanol.
Method:
- Transformation: Transform the plasmid into chemically competent SHuffle cells. Plate on LB-agar with antibiotic. Incubate at 30°C for 24-48 hours.
- Inoculum: Pick a single colony into 5 mL LB+antibiotic. Grow overnight at 30°C, 220 rpm.
- Expression Culture: Dilute overnight culture 1:100 into fresh TB+antibiotic (e.g., 50 mL in a 250 mL baffled flask). Grow at 30°C until OD600 ~0.5-0.6.
- Induction: Add IPTG to a final concentration of 0.1 mM. Reduce temperature to 16°C. Continue incubation for 16-20 hours.
- Harvest: Pellet cells at 4,000 x g for 20 min at 4°C. Discard supernatant. Cell pellet can be stored at -80°C.
- Lysis: Thaw pellet on ice. Resuspend in cold Lysis Buffer (5 mL per gram pellet). Incubate on ice for 30 min. Sonicate on ice (10 cycles of 30 sec on/30 sec off). Clarify lysate by centrifugation at 15,000 x g for 30 min at 4°C.
- Analysis:
- Solubility Check: Analyze supernatant (soluble) and pellet (insoluble) fractions by SDS-PAGE.
- Disulfide Check: Run identical samples on non-reducing (no DTT/β-ME in sample buffer) and reducing (with DTT/β-ME) SDS-PAGE. A band shift indicates disulfide formation.
Visualizing the Engineered Cytoplasmic Redox Pathway
Diagram Title: Engineered E. coli Cytoplasmic Disulfide Pathway
Experimental Workflow for Cytoplasmic Production
Diagram Title: Cytoplasmic Disulfide Protein Production Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Research |
|---|---|
| SHuffle T7 Express E. coli | Engineered strain with oxidizing cytoplasm (ΔtrxB gor ahpC) and cytoplasmic DsbC for disulfide bond formation and isomerization. |
| pET Expression Vectors | High-copy number plasmids with strong T7 promoter for controllable, high-level protein expression. |
| Terrific Broth (TB) Media | Nutrient-rich formulation supporting high cell density, often leading to higher recombinant protein yields. |
| Protease Inhibitor Cocktail (for E. coli) | Protects target proteins from degradation by endogenous proteases during cell lysis and purification. |
| GSH/GSSG Redox Buffer | Glutathione redox pair used to fine-tune the redox potential in in vitro refolding or cell culture experiments. |
| Lysozyme | Enzyme that degrades the bacterial cell wall, essential for efficient lysis while maintaining protein integrity. |
| Non-Reducing SDS-PAGE Sample Buffer | Contains SDS but no DTT/β-ME, allowing analysis of protein migration with intact disulfide bonds. |
Technical Support Center: Troubleshooting & FAQs for Cytoplasmic Disulfide Bond Formation inE. coli
Context: This support center addresses common challenges encountered while implementing strategies from the thesis "Enhancing disulfide bond formation in E. coli cytoplasm" for the production of disulfide-rich therapeutic proteins.
Frequently Asked Questions (FAQs)
Q1: My target protein (e.g., scFv antibody fragment) is expressed in the cytoplasm but is largely insoluble and inactive. What are the primary system components I should verify? A: This indicates inadequate redox control. Verify the following:
- Host Strain: Confirm you are using a suitable E. coli strain with mutations in the reducing pathways (e.g., trxB / gor double mutant like SHuffle). In SHuffle T7 Express, the thioredoxin reductase (trxB) and glutathione reductase (gor) genes are knocked out.
- Disulfide Bond Isomerase: Ensure co-expression of a disulfide bond isomerase like DsbC (which is typically expressed in the cytoplasm in engineered strains) to correct mis-formed bonds.
- Expression Conditions: Use lower growth temperatures (e.g., 25-30°C) to slow protein synthesis and favor folding.
Q2: Despite using an engineered strain, my yield of active growth factor (e.g., β-NGF) is low. How can I optimize the expression protocol? A: Low yield of active product often relates to culture conditions.
- Induction Parameters: Reduce the inducer concentration (e.g., IPTG to 0.05-0.1 mM) and induce at a lower optical density (OD~0.4-0.6).
- Aeration: Ensure high aeration during post-induction, as oxygen is required for disulfide bond formation.
- Harvest Time: Perform a time course; harvest between 8-16 hours post-induction at low temperature to minimize aggregation.
Q3: I observe excessive protein aggregation even with a trxB/gor mutant strain. What further genetic or process modifications can I try? A: Aggregation suggests folding is still overwhelmed. Consider:
- Fusion Tags: Utilize a soluble fusion partner tag (e.g., MBP, SUMO, DsbC itself) to enhance initial solubility, followed by cleavage.
- Co-expression of Chaperones: Co-express cytoplasmic chaperone systems like GroEL/GroES or DnaK/DnaJ/GrpE to assist folding.
- Target-Specific Optimization: Some disulfide-rich proteins (e.g., defensins) may require very specific N-terminal sequences or leader peptides for optimal folding; consult literature for your specific biologic.
Q4: How do I quantitatively assess the improvement in disulfide bond formation in my experiments? A: Use these analytical techniques:
- Activity Assay: Compare specific activity (units/mg) of the purified protein to a standard.
- Electrophoresis: Use non-reducing vs. reducing SDS-PAGE. A faster migration under non-reducing conditions often indicates a more compact, disulfide-bonded structure.
- Mass Spectrometry: Confirm the molecular weight matches the oxidized form.
- Chromatography: Analyze purity and correct folding via Reverse-Phase HPLC or Analytical Size-Exclusion Chromatography.
Q5: What are the critical control experiments when benchmarking a new system for producing a disulfide-rich biologic? A: Always run parallel controls:
- Expression of the same construct in a standard, non-engineered E. coli strain (e.g., BL21(DE3)).
- Expression of a well-characterized, disulfide-bonded positive control protein (e.g., Bovine Pancreatic Trypsin Inhibitor - BPTI) in your experimental strain.
- Expression of a catalytically inactive mutant of your target protein (if applicable) to distinguish between expression and folding issues.
Experimental Protocols
Protocol 1: Small-Scale Expression Test for Disulfide-Rich Proteins in E. coli SHuffle Strains
Objective: To screen for soluble, active expression of a target protein (e.g., Fab antibody fragment).
Materials:
- SHuffle T7 Express E. coli competent cells (or equivalent ΔtrxB Δgor ssrA* mutant).
- Plasmid containing target gene (optimized for E. coli, with periplasmic signal sequence removed) in a T7 or compatible vector.
- Plasmid for DsbC co-expression (if not already chromosomally encoded).
- LB medium with appropriate antibiotics (e.g., 100 µg/mL ampicillin, 34 µg/mL chloramphenicol).
- IPTG (Isopropyl β-D-1-thiogalactopyranoside).
- Lysis Buffer: 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mg/mL Lysozyme, 0.1% Triton X-100, 1 mM PMSF, 5 mM EDTA.
- Benzonase Nuclease (optional).
Method:
- Transform plasmids into SHuffle cells. Plate on selective agar. Incubate at 30°C for 24-36 hours.
- Inoculate 3-5 mL of selective LB medium with a single colony. Grow overnight at 30°C, 220 rpm.
- Dilute the overnight culture 1:100 into 25 mL fresh selective LB in a 125 mL baffled flask. Grow at 30°C until OD600 reaches 0.5-0.6.
- Induce protein expression by adding IPTG to a final concentration of 0.1 mM.
- Incubate post-induction at 30°C for 16-20 hours (or 25°C for 24 hours) with shaking.
- Harvest cells by centrifugation at 4,000 x g for 15 min at 4°C.
- Resuspend cell pellet in 2-3 mL Lysis Buffer. Incubate on ice for 30 min.
- Lyse cells by sonication (5 cycles of 30 sec pulse, 30 sec rest on ice) or by freeze-thaw.
- Clarify the lysate by centrifugation at 16,000 x g for 30 min at 4°C.
- Separate supernatant (soluble fraction) from pellet (insoluble fraction).
- Analyze both fractions by SDS-PAGE (under reducing and non-reducing conditions) and perform an activity assay if available.
Protocol 2: Non-Reducucing vs. Reducing SDS-PAGE Analysis
Objective: To rapidly assess disulfide bond formation in expressed protein.
Method:
- Prepare two identical samples of your protein lysate or purified sample.
- To Sample A (Reducing), add 1X Laemmli buffer containing 5% β-mercaptoethanol (or 20 mM DTT). Heat at 95°C for 5-10 minutes.
- To Sample B (Non-Reducing), add 1X Laemmli buffer without any reducing agent. Do not heat, or heat at a lower temperature (e.g., 37°C for 10 min) to avoid heat-induced aggregation.
- Load both samples on the same polyacrylamide gel (4-20% gradient gel recommended).
- Run electrophoresis.
- Compare migration. A protein with intact disulfide bonds will typically migrate faster in the non-reducing lane (Sample B) due to a more compact structure, compared to its reduced, linearized form in Sample A.
Table 1: Comparison of E. coli Strains for Cytoplasmic Production of Disulfide-Rich Proteins
| Strain Genotype | Key Feature | Typical Application | Reported Yield Range for Model Proteins* | Major Advantage | Major Limitation |
|---|---|---|---|---|---|
| BL21(DE3) | Wild-type redox cytoplasm | Control for insoluble expression | <1 mg/L (active, for disulfide proteins) | Robust growth, high biomass | Highly reducing cytoplasm |
| Origami(DE3) | ΔtrxB Δgor mutant | Cytoplasmic expression | 5-50 mg/L | Strongly oxidizing cytoplasm | Slow growth, prone to aggregation |
| SHuffle T7 | ΔtrxB Δgor ssrA cytoplasmic DsbC | Cytoplasmic expression & folding | 10-100 mg/L | Combines oxidizing cytoplasm with isomerase activity | Slower growth than wild-type |
| C43(DE3) pLysS | Membrane mutation | Difficult membrane proteins | Varies widely | Tolerates toxic proteins | Not specifically oxidizing |
*Yields are highly protein-dependent. Model proteins include scFv, Fab, BPTI, etc. Data compiled from recent literature and vendor specifications.
Table 2: Troubleshooting Guide: Symptoms, Causes, and Solutions
| Symptom | Possible Cause | Recommended Solution |
|---|---|---|
| No protein expression | Poor transformation, wrong strain/plasmid, toxic protein | Verify plasmid sequence, use tighter promoter (e.g., pBad), test in non-T7 strain, use lower copy number vector. |
| Protein only in insoluble fraction | Aggregation due to rapid synthesis, lack of chaperones, incorrect redox | Lower induction temperature (25°C), reduce IPTG concentration, co-express GroEL/GroES, use fusion tag. |
| Protein soluble but inactive | Misfolding, incorrect disulfide pairing | Ensure DsbC (isomerase) is present, optimize redox buffer during lysis, screen different strains. |
| Low overall yield | Protein degradation, poor growth | Use protease-deficient strain (e.g., lon ompT), increase culture medium richness, optimize harvest time. |
| Multiple bands on gel | Proteolysis, incomplete disulfide formation | Add protease inhibitor cocktail, test different lysis buffers, check for periplasmic leakage. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Cytoplasmic Disulfide Bond Formation Experiments
| Item | Function | Example/Description |
|---|---|---|
| Engineered E. coli Strain | Provides oxidizing cytoplasm and folding catalysts. | SHuffle series (NEB), Origami series (Novagen), or equivalent ΔtrxB Δgor mutants with DsbC. |
| Expression Vector | Drives controlled, high-level protein expression. | pET series (with T7 promoter), pBAD (arabinose-inducible for tight control). |
| Fusion Tag System | Enhances solubility, enables purification. | His-tag (IMAC purification), MBP (maltose-binding protein), SUMO (aids solubility and cleavage). |
| Disulfide Isomerase Plasmid | Co-expresses catalyst for correct bond pairing (if not in host genome). | Plasmid encoding DsbC (or PDI for more complex proteins). |
| Chaperone Plasmid Set | Co-expresses folding assistants to prevent aggregation. | Plasmids for GroEL/GroES or DnaK/DnaJ/GrpE systems. |
| Activity Assay Kit | Quantifies functional protein yield. | ELISA kits (for antibodies), enzyme-specific substrate kits (for kinases/growth factors). |
| Redox Buffering Additives | Maintains redox state during cell lysis and purification. | Cysteine/Cystine pair, Reduced/Oxidized Glutathione (GSH/GSSG). |
Visualizations
Title: Engineering E. coli for Cytoplasmic Disulfide Bond Formation
Title: Optimized Workflow for Cytoplasmic Expression
Engineering Solutions: Practical Strategies to Create an Oxidation-Friendly Cytoplasm in E. coli
Technical Support Center: Troubleshooting & FAQs
Frequently Asked Questions
Q1: I have constructed an E. coli ΔtrxB/Δgor double mutant, but my target protein with multiple disulfide bonds is still not folding properly. What could be wrong? A: While knocking out the major reductive pathways (trxB, gor) is foundational, several other factors can interfere:
- Cysteine Residue Accessibility: The cysteines in your target protein may be buried or mispositioned, preventing correct pairing. Consider structural analysis or directed evolution.
- Thioredoxin 2 (trxC) or Glutaredoxin Systems: These parallel reductive systems may still be active. Consider adding a trxC mutation or using strains with additional knockouts (e.g., ahpC).
- Expression Conditions: Overly rapid expression can lead to aggregation. Reduce inducer concentration (e.g., IPTG to 0.01-0.1 mM), lower temperature (25-30°C), or use a weaker promoter.
- Chaperone Limitation: Co-expression of disulfide bond isomerases like DsbC (which requires DsbD for reduction in the cytoplasm) can be crucial for complex proteins.
Q2: My double mutant strain grows extremely slowly or is non-viable. How can I improve viability? A: The trxB gor double knockout creates a strong oxidative stress. These are standard remediation steps:
- Supplement Media: Add 0.5-2 mM reduced cysteine or 1-5 mM DTT to the growth medium to provide a reductive boost.
- Use Rich Media: Grow cultures in rich media (e.g., 2xYT) rather than minimal media to provide more metabolic resources.
- Strain Validation: Ensure you are using a strain with compensatory mutations, such as ahpC or suppressor mutations, which are present in commercial strains like SHuffle T7. Do not attempt to construct the double mutant in a wild-type background.
- Aeration Control: Moderate shaking is preferable; excessive aeration can increase oxidative stress.
Q3: I am getting high levels of protein aggregation in my mutant strain. What optimization strategies should I try? A: Aggregation indicates non-native interactions. Follow this protocol:
- Screen Expression Parameters: Set up a matrix of inducer concentration (IPTG: 0.01, 0.05, 0.1 mM) vs. temperature (20°C, 25°C, 30°C).
- Solubility Check: Analyze whole cell lysate (W), soluble fraction (S), and insoluble pellet (P) by SDS-PAGE.
- Chaperone Co-expression: Test co-expression vectors for chaperone pairs (GroEL/GroES, DnaK/DnaJ/GrpE) or DsbC/DsbD.
- Purification under Denaturing Conditions: If aggregation persists, purify from the inclusion body pellet using urea or guanidine HCl, followed by in vitro refolding.
Q4: How do I verify that the reductive pathways are successfully knocked out in my strain? A: Use a functional assay. The Disulfide Bond Reporter Assay is standard:
- Principle: Express a reporter protein (e.g., mouse urokinase, alkaline phosphatase) that requires disulfide bonds for activity. Activity in the cytoplasm indicates a functional oxidative environment.
- Protocol: Transform the reporter plasmid into your mutant and a wild-type control. Grow cultures, prepare periplasmic and cytoplasmic fractions separately. Measure reporter enzyme activity in each fraction. In a successful trxB/gor mutant, cytoplasmic activity should be significantly higher than in the wild-type.
Q5: What are the key differences between commercial trxB/gor mutant strains (e.g., SHuffle, Origami) and which should I choose? A: See the comparison table below.
Table 1: Comparison of Common E. coli Strains for Cytoplasmic Disulfide Bond Formation
| Strain Genotype (Key Mutations) | Commercial Example | Cytoplasmic Redox Environment | Typical Growth Rate | Key Best Use Case | Compensatory Mutations |
|---|---|---|---|---|---|
| Wild-Type (e.g., BL21) | BL21(DE3) | Strongly Reducing | Fast (+++) | Standard soluble proteins, no disulfides | None |
| ΔtrxB (Thioredoxin Reductase) | Origami B(DE3) | Oxidizing | Slow (+) | Proteins with simple disulfides | gor (Glutathione Reductase) |
| Δgor (Glutathione Reductase) | - | Mildly Oxidizing | Moderate (++) | Rarely used alone | May require trxB |
| ΔtrxB Δgor | Origami 2(DE3), SHuffle T7 | Highly Oxidizing | Very Slow (+/-) | Complex, multi-disulfide proteins | ahpC (peroxiredoxin) in SHuffle |
| ΔtrxB Δgor ΔahpC + dsbC (cytoplasmic) | SHuffle T7 Express | Optimized Oxidizing + Isomerization | Slow (+) | Challenging proteins requiring isomerization | Genotype stabilized |
Table 2: Troubleshooting Matrix for Low Protein Yield in trxB/gor Mutants
| Symptom | Possible Cause | Diagnostic Test | Solution |
|---|---|---|---|
| No protein expression | Plasmid loss, toxicity | Plate on selective antibiotic, check plasmid stability | Use stricter antibiotic selection, lower copy number vector |
| Protein in inclusion bodies | Aggregation due to fast folding/oxidation | Solubility fractionation (SDS-PAGE) | Lower expression temperature (20-25°C), reduce inducer, add chaperones |
| Low cell density at harvest | Strain sensitivity, media issue | Measure OD600 over time | Use rich media (2xYT), add 0.5-1 mM cysteine, reduce aeration |
| Protein degradation | Protease activity | Add protease inhibitors (PMSF) to lysis buffer | Use protease-deficient strain (e.g., lon ompT), shorten induction time |
Experimental Protocols
Protocol 1: Functional Validation of Reductive Pathway Knockout using Alkaline Phosphatase (PhoA) Activity Assay Objective: Confirm the oxidative cytoplasmic environment in your trxB/gor mutant. Reagents: pTA-PhoA plasmid (PhoA without signal sequence), pTrc99a vector, 1M Tris-HCl (pH 8.0), 0.1M p-Nitrophenyl Phosphate (pNPP), 2M NaOH. Steps:
- Transform the pTA-PhoA plasmid and an empty vector control into your mutant strain and a wild-type control (e.g., BL21).
- Grow cultures in LB+Amp at 30°C to mid-log phase (OD600 ~0.6). Induce with 1 mM IPTG for 2 hours.
- Harvest 1 mL of cells. Prepare cytoplasmic fraction via osmotic shock or lysozyme/EDTA treatment followed by centrifugation.
- In a 96-well plate, mix 50 µL of cytoplasmic extract with 150 µL of assay buffer (1M Tris-HCl, pH 8.0).
- Start reaction by adding 50 µL of 0.1M pNPP. Incubate at 37°C for 10-30 min.
- Stop reaction with 50 µL of 2M NaOH. Measure absorbance at 405 nm.
- Interpretation: A significantly higher A405 in the mutant's extract vs. wild-type confirms a more oxidizing cytoplasm.
Protocol 2: Optimizing Expression for Soluble Yield in trxB/gor Mutants Objective: Find conditions that maximize soluble expression of a disulfide-bonded target protein. Reagents: Expression plasmid, IPTG (varying concentrations), 2xYT media, Lysis Buffer (PBS, pH 7.4, 1 mg/mL lysozyme, protease inhibitors). Steps:
- Inoculate 5 mL overnight cultures in 2xYT with antibiotic.
- Dilute 1:100 into fresh 2xYT (10 mL cultures in 50 mL flasks). Grow at 30°C to OD600 ~0.6.
- Induce using an IPTG gradient (e.g., 0.01, 0.05, 0.1, 0.5 mM). Split each induced culture into two flasks.
- Incubate one set at 30°C and the other at 20°C for 16-18 hours post-induction.
- Harvest cells by centrifugation. Resuspend pellet in 1 mL Lysis Buffer. Incubate 30 min on ice, then sonicate.
- Centrifuge lysate at 15,000 x g for 30 min at 4°C to separate soluble (supernatant) and insoluble (pellet) fractions.
- Analyze equal proportions of total lysate (T), soluble (S), and resuspended pellet (P) by SDS-PAGE.
Visualizations
Diagram 1: E. coli Cytoplasmic Redox Pathways in WT vs trxB/gor Mutant
Diagram 2: Experimental Workflow for Protein Expression in trxB/gor Strains
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents for Working with trxB/gor E. coli Mutants
| Item | Function & Rationale | Example/Notes |
|---|---|---|
| Specialized E. coli Strains | Provide the genetically engineered oxidative cytoplasm background. Essential starting point. | SHuffle T7 Express (C3029J), Origami 2(DE3) (Novagen). |
| Redox Media Supplements | Improve viability of sensitive double mutants by providing reducing power. | Dithiothreitol (DTT, 1-5 mM), Reduced L-Cysteine (0.5-2 mM). |
| Chaperone/Isomerase Plasmid | Co-expression vectors to assist folding and correct disulfide isomerization. | pTf16 (Takara, for TF), pG-KJE8 (for DnaK/GrpE), pDsbC (for DsbC). |
| Disulfide Bond Reporter | Functional validation of cytoplasmic oxidation. Plasmids expressing disulfide-dependent enzymes. | pTA-PhoA (alkaline phosphatase), pET-22b-Δss-scFv (antibody fragment). |
| Low-IPTG or Autoinduction Media | Enables slow, controlled protein expression to minimize aggregation. | Overnight Express Autoinduction System (Novagen), or prepare custom low-IPTG media. |
| Protease Inhibitor Cocktail | Protects target proteins from degradation, especially in stressed mutant strains. | EDTA-free cocktails (e.g., Roche cOmplete) are often preferred for metalloproteinases. |
| Non-Reducing SDS-PAGE Sample Buffer | Allows analysis of disulfide-bonded states without reducing all bonds before electrophoresis. | Sample buffer without β-mercaptoethanol or DTT. |
| Mass Spectrometry Reagents | For definitive confirmation of disulfide bond formation and mapping. | Iodoacetamide (alkylation), Trypsin/Lys-C (digestion), TCEP (reducing agent for MS). |
Technical Support Center
Troubleshooting & FAQ
Q1: My co-expression of sulfhydryl oxidase (Erv1p) and disulfide bond isomerase (DsbC) in the E. coli cytoplasm is not improving the yield of active recombinant protein. What could be wrong? A: This is often due to insufficient redox equilibrium. Ensure you are also co-expressing a reductase (e.g., E. coli TrxA/B or yeast NADPH oxidase) to recycle the oxidase and prevent hyperoxidation. Check the culture medium; supplementing with 1-5 mM cysteine can help maintain a poise. Verify plasmid compatibility and promoter strength—unequal expression levels are common. Run a Western blot to confirm both catalysts are present.
Q2: I observe significant cell lysis or growth retardation upon induction of the oxidative catalyst genes. How can I mitigate this? A: Cytoplasmic expression of these catalysts increases oxidative stress. Use a tightly regulated promoter (e.g., T7, pBAD) and titrate the inducer concentration (e.g., 0.01-0.5 mM IPTG). Consider using an E. coli strain with enhanced oxidative stress resistance (e.g., trxB/gor mutants, SHuffle strains). Lower the incubation temperature post-induction (25-30°C) and monitor OD600 closely. Pre-induction viability should be >0.8.
Q3: My disulfide-bonded protein aggregates in inclusion bodies even with co-expression. What optimization steps should I take? A: Aggregation indicates folding is outcompeted. Implement a sequential expression protocol: induce the catalyst genes first for 1-2 hours, then induce the target protein. Decrease induction temperature to 20-25°C. In your lysis buffer, include 1-2 mM NEM (N-ethylmaleimide) to alkylate free thiols and "snapshot" the redox state. Screen different sulfhydryl oxidase/disulfide isomerase pairs (e.g., Erv1p/Pdi, ALR/ALR).
Q4: How do I accurately measure the in vivo redox state of my target protein to confirm catalyst activity? A: Use the alkylation trapping assay. Harvest cells rapidly into cold 10% TCA to freeze metabolism. Pellet, wash with acetone, and lyse. Resuspend the pellet in a buffer with iodoacetamide (IAM) to alkylate free thiols, or first reduce with DTT then alkylate with 4-acetamido-4'-maleimidylstilbene-2,2'-disulfonic acid (AMS), which adds ~0.5 kDa per free thiol. Analyze by non-reducing SDS-PAGE. A mobility shift indicates the number of disulfide bonds formed.
Q5: My disulfide isomerase (DsbC) appears inactive. What controls should I run? A: DsbC requires a dimeric state and its own N-terminal disulfide for chaperone activity. Check reducing agent in buffers; keep <0.1 mM DTT. Confirm dimerization via non-reducing SDS-PAGE (it runs at ~50 kDa). A positive control: assess its ability to refold scrambled RNase A in vitro. For in vivo, co-express with a model disulfide-rich protein (e.g., bovine pancreatic trypsin inhibitor) as a benchmark.
Table 1: Common Sulfhydryl Oxidase & Disulfide Isomerase Pairs for E. coli Cytoplasm
| Oxidase | Isomerase/Chaperone | Key Features | Typical Fold Improvement | Optimal Strain |
|---|---|---|---|---|
| Yeast Erv1p | Yeast Pdi1p | Complete eukaryotic system; requires FAD & heme. | 5-20x (varies by target) | Origami B(DE3) (trxB/gor) |
| Human ALR | Human PDI | Shorter, cytosolic version of ALR often used (hALR_c). | 3-15x | SHuffle T7 (trxB/gor, dsbC periplasm) |
| E. coli DsbA (cytosolic mutant) | E. coli DsbC (cytosolic) | Prokaryotic pair; can bypass need for redox cofactors. | 2-8x | BL21(DE3) ΔdsbA |
| V. cholerae Ero1 | V. cholerae DsbC | Highly efficient but can be toxic; tight control needed. | Up to 25x (reported) | Custom trxB/gor/ahpC mutant |
Table 2: Troubleshooting Metrics for Common Problems
| Problem | Probable Cause | Diagnostic Assay | Typical Target Value for Optimization |
|---|---|---|---|
| Low soluble yield | Hyperoxidation, Aggregation | Soluble/Insoluble fractionation, AMS assay | Soluble fraction >30% of total target protein |
| No catalytic activity | Misfolded disulfides, inactive enzyme | In vitro activity assay (e.g., RNase refolding), Non-red. PAGE | Catalytic turnover >1 min⁻¹ for control substrate |
| High cell death | Oxidative stress, toxicity | Viability plating (CFU/mL), ROS staining (DCFDA) | Post-induction viability >70% vs uninduced |
| Inconsistent results | Plasmid instability, uneven expression | Plasmid retention assay, qRT-PCR | >95% plasmid retention, mRNA ratio 1:1:1 (Ox:Isom:Target) |
Experimental Protocols
Protocol 1: Sequential Induction for Cytoplasmic Disulfide Bond Formation
- Transform E. coli SHuffle T7 cells with two compatible plasmids: one expressing the sulfhydryl oxidase/disulfide isomerase pair (e.g., pETDuet-erv1p-pdi1), and another expressing the target protein (e.g., pCOLADuet-target).
- Inoculate 5 mL LB with both antibiotics. Grow overnight at 30°C, 220 rpm.
- Dilute 1:100 into fresh TB medium with antibiotics. Grow at 30°C to OD600 ~0.6.
- First Induction: Add IPTG to 0.1 mM to induce the catalyst genes. Incubate at 30°C for 2 hours.
- Second Induction: Add IPTG to a final concentration of 0.5 mM to induce the target protein. Shift temperature to 25°C. Incubate for 16-20 hours.
- Harvest cells by centrifugation (4,000 x g, 20 min). Process for protein purification or analysis.
Protocol 2: Alkylation Trapping Assay for Redox State Analysis
- Quench: At harvest, mix 1 mL culture directly with 0.1 mL of 100% (w/v) ice-cold TCA. Incubate on ice for 30 min.
- Pellet: Centrifuge at 16,000 x g, 15 min, 4°C. Wash pellet twice with 1 mL ice-cold acetone. Air dry.
- Alkylate (Option A - Free Thiols): Resuspend pellet in 50 µL denaturing lysis buffer (6 M Guanidine-HCl, 100 mM Tris, 1 mM EDTA, pH 8.0) with 20 mM IAM. Incubate in dark, 30°C, 30 min.
- Alkylate (Option B - Total Thiols): Resuspend in 50 µL denaturing lysis buffer with 10 mM DTT. Incubate 30°C, 30 min. Add 40 mM AMS, incubate in dark, 30°C, 2 hours.
- Analyze: Add SDS-PAGE loading buffer (without reducing agent). Boil 5 min. Run on 12-15% non-reducing SDS-PAGE. Western blot for target protein.
Diagrams
Title: Cytoplasmic Disulfide Bond Formation Catalysis Pathway
Title: Sequential Induction Experimental Workflow
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Reagent/Material | Function/Benefit | Example & Notes |
|---|---|---|
| SHuffle T7 Express E. coli | Engineered for cytoplasmic disulfide bonds. trxB/gor mutations enhance catalyst activity. | NEB C3026J. Constitutively expresses DsbC in periplasm, but used for cytosolic work. |
| pETDuet-1 Vector | Co-expression of two genes (e.g., oxidase & isomerase) from a single plasmid with T7 promoters. | EMD Millipore, 71146-3. Allows controlled stoichiometry. |
| Anti-"Tag" Antibodies | Essential for detecting catalysts and target via Western blot amid high stress protein loads. | Anti-His, Anti-FLAG, Anti-HA. Confirm expression levels. |
| AMS (4-Acetamido-4'-maleimidylstilbene-2,2'-disulfonic acid) | Thiol-alkylating agent that causes a quantifiable gel shift for each free cysteine. | Thermo Fisher, A485. Use fresh, protect from light. |
| NEM (N-Ethylmaleimide) | Rapid, membrane-permeable alkylating agent for in vivo trapping of free thiols at harvest. | Sigma-Aldrich, E3876. Prepare fresh in ethanol. |
| FAD & Heme Precursors | Cofactors for some sulfhydryl oxidases (e.g., Erv1p). Supplementing can boost activity. | Add 10 µM FAD and 5 µM Hemin to culture at induction. |
| Tunable Autoinduction Media | Provides consistent growth into stationary phase with automatic induction; improves reproducibility. | Formulations with lactose/glycerol for T7 systems. |
| Catalase | Co-express or add to medium to degrade H₂O₂ produced by oxidases, reducing cellular stress. | E. coli KatG or bovine liver catalase (add 100 U/mL). |
Technical Support Center: Troubleshooting & FAQs
Frequently Asked Questions
Q1: My target protein is expressed in SHuffle but remains entirely insoluble. What are the primary troubleshooting steps? A1: Begin by confirming the redox environment. SHuffle's cytoplasm is oxidizing, but if expression is too rapid, misfolding can still occur. Implement the following protocol:
- Reduce Expression: Lower incubation temperature to 16-25°C post-induction and reduce inducer (e.g., IPTG) concentration to 0.01-0.1 mM.
- Evaluate Solubility: Perform small-scale expression tests with varying induction times (2, 4, 6 hours). Lyse cells using BugBuster or sonication in a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA. Centrifuge at 15,000 x g for 20 min. Analyze supernatant (soluble) and pellet (insoluble) fractions by SDS-PAGE.
- Co-expression: Co-express with a chaperone plasmid (e.g., pGro7 for GroEL/ES or pTf16 for DnaK/DnaJ/GrpE) supplied with the SHuffle strain kit.
Q2: The Origami strain exhibits very slow growth. Is this normal, and how do I compensate? A2: Yes, slower growth is characteristic due to mutations in both trxB and gor genes, which impair the thioredoxin and glutathione reductase pathways. Compensate by:
- Using richer media (2xYT or Terrific Broth instead of LB).
- Extending pre-culture and main culture growth times. Ensure cultures are thoroughly acclimated to antibiotic selection (tetracycline and kanamycin) by streaking from a fresh glycerol stock onto selective plates.
- Inoculating main cultures at a higher starting OD600 (e.g., 0.05-0.1) from a dense pre-culture.
Q3: With CyDisCo, I see improper disulfide bonding. How can I verify the pattern and optimize conditions? A3: Improper pairing requires analytical verification and system tuning.
- Verification Protocol: Perform non-reducing vs. reducing SDS-PAGE. A faster mobility shift under non-reducing conditions indicates intramolecular disulfide formation. For precise mapping, use mass spectrometry (MS) after tryptic digest under non-reducing conditions.
- Optimization: Titrate the co-expressed catalysts (DsbC and/or PDI variants). Use varying concentrations of arabinose inducer (e.g., 0.002%, 0.02%, 0.2%) for the pACYC-derived plasmid carrying the catalyst genes. Ensure the main target protein is on a separate, compatible plasmid (e.g., pET-based) with a different inducer (IPTG).
Q4: What is the critical difference in antibiotic selection between these strains, and what is a common mistake? A4: Each strain has unique selection markers essential for maintaining its genotype. A common mistake is using incomplete or incorrect antibiotic cocktails.
Table 1: Strain Genotypes and Required Antibiotics
| Strain | Key Genotype Modifications | Required Antibiotics for Maintenance | Typical Working Concentrations |
|---|---|---|---|
| SHuffle | Δ(gor) Δ(trxB), ahpC*, *dsbC (periplasmic) | Chloramphenicol, Kanamycin | 34 µg/mL, 50 µg/mL |
| Origami | Δ(gor) Δ(trxB) | Tetracycline, Kanamycin | 12.5 µg/mL, 15 µg/mL |
| CyDisCo | (No inherent chromosomal resistance; conferred by plasmids) | Depends on plasmid(s). Common: Ampicillin (Target), Chloramphenicol (Catalyst) | 100 µg/mL, 34 µg/mL |
Q5: How do I choose between these three systems for a new protein? A5: Base your initial choice on the complexity and localization of disulfide bonds within your target protein. Table 2: System Selection Guide
| System | Optimal Use Case | Oxidizing Engine | Typical Yield Range (Soluble, mg/L) | Key Limitation |
|---|---|---|---|---|
| SHuffle | Proteins with 1-2 disulfides; cytoplasmic expression. | Mutated AhpC*, DsbC in cytoplasm. | 5 - 50 mg/L | Can struggle with complex/multiple disulfides. |
| Origami | Proteins requiring a more strongly oxidizing cytoplasm than SHuffle. | Combined trxB/gor mutations. | 1 - 20 mg/L | Very slow growth, lower biomass yield. |
| CyDisCo | Complex eukaryotic proteins with multiple/nested disulfides. | Co-expressed eukaryotic PDI and bacterial DsbC. | 0.5 - 10 mg/L | Requires careful tuning of catalyst-to-target ratio. |
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents
| Reagent/Material | Function | Example Product/Catalog # |
|---|---|---|
| BugBuster HT Protein Extraction Reagent | Gentle, non-denaturing lysis for solubility analysis. | MilliporeSigma, 70922 |
| Lysozyme | Enzymatic cell wall degradation for efficient lysis. | MillipopreSigma, L6876 |
| Tris(2-carboxyethyl)phosphine (TCEP) | Strong, thiol-specific reducing agent for reducing SDS-PAGE controls. | Thermo Fisher, 20490 |
| 4-Acetamido-4'-maleimidylstilbene-2,2'-disulfonic acid (AMS) | Thiol-alkylating agent for trapping free cysteines, assessing oxidation state via gel shift. | Thermo Fisher, A485 |
| pGro7 Chaperone Plasmid | Co-expression of GroEL/ES chaperonins to assist folding. | Takara Bio, 3340 |
| Arabinose (low concentration grade) | Precise induction of CyDisCo catalyst expression. | GoldBio, A-300 |
Experimental Protocols
Protocol 1: Initial Solubility Screening for Disulfide-Bonded Proteins
- Transformation: Transform your target plasmid into SHuffle, Origami, and a control strain (e.g., BL21(DE3)).
- Inoculation: Pick 3-5 colonies into 5 mL LB with appropriate antibiotics. Grow overnight at 30°C (SHuffle, Origami) or 37°C (control).
- Induction: Dilute cultures 1:100 into fresh medium + antibiotics. Grow to OD600 ~0.6-0.8. Induce with optimal IPTG concentration (e.g., 0.1 mM for SHuffle/Origami, 0.5 mM for control). Incubate at 25°C for 16-20 hours.
- Lysis: Harvest 1 mL culture. Resuspend pellet in 100 µL BugBuster reagent with 1 µL Benzonase and Lysozyme (1 kU). Incubate on rotator for 20 min at RT.
- Fractionation: Centrifuge at 15,000 x g for 20 min. Transfer supernatant (soluble). Wash pellet with 100 µL PBS, re-centrifuge, discard wash, and resuspend pellet in 100 µL PBS + 1% SDS (insoluble).
- Analysis: Run 20 µL of each fraction on reducing and non-reducing SDS-PAGE.
Protocol 2: Verifying Disulfide Bond Formation via Non-Reducing SDS-PAGE
- Prepare protein samples in non-reducing Laemmli buffer (no β-mercaptoethanol or DTT). Include a parallel sample with added TCEP (10 mM final) as a reduced control.
- Do not boil samples for the non-reduced set, as heat can cause scrambling. Incubate at 37°C for 15 minutes. Boil the reduced control sample as usual.
- Load and run the gel using standard SDS-PAGE procedures.
- Compare mobility. A properly oxidized protein with intramolecular disulfides will migrate faster than its reduced (linearized) form.
System Workflow Diagrams
SHuffle Strain Expression and Analysis Workflow
CyDisCo Two-Plasmid Co-expression Optimization
Fusion Tags and Chaperone Co-expression to Assist Folding and Solubility
Technical Support Center
Troubleshooting Guides & FAQs
Q1: I am expressing a target protein with a disulfide bond in the E. coli cytoplasm using a fusion tag (e.g., MBP, Trx). The solubility is improved, but the protein is mostly in the reduced, inactive form. What can I do?
A: This is common. The cytoplasmic environment is reducing. To promote disulfide bond formation, you must engineer the cytoplasm. Use an E. coli strain genetically modified to enhance disulfide bond formation, such as SHuffle T7 Express. Co-express a catalyst for disulfide bond formation. The most common and effective system is co-expression of the DsbC chaperone, which catalyzes isomerization and correction of mispaired bonds.
- Protocol: Co-transform your fusion-tag expression vector with a plasmid expressing DsbC under its own promoter (e.g., pACYC184-based). Induce DsbC expression 1 hour prior to inducing your target protein. Ensure the medium contains a low level of a redox agent like cysteine (e.g., 5 mM) to support the Dsb pathway.
Q2: My target protein is insoluble even when fused to a solubility-enhancing tag like SUMO or MBP in a standard BL21(DE3) strain. What's the next step?
A: The fusion tag may not be sufficient alone. Implement a combined strategy:
- Switch to a folding-enhanced strain: Use SHuffle or Origami B (which has mutations in both thioredoxin reductase and glutathione reductase).
- Co-express a cytoplasmic chaperone pair: The GroEL-GroES (Hsp60/Hsp10) system is crucial for folding many proteins. Co-expression can prevent aggregation.
- Optimize expression conditions: Lower the induction temperature (to 18-25°C), reduce IPTG concentration (to 0.1-0.5 mM), and extend post-induction time.
- Protocol for GroEL/GroES co-expression: Use a compatible plasmid (e.g., pGro7 from Takara Bio) expressing the groEL-groES operon. Induce chaperone expression with L-arabinose (0.5 mg/mL) at least 30 minutes before target protein induction.
Q3: After successful expression and folding, how do I remove the fusion tag without disrupting the formed disulfide bonds?
A: Choose a protease that is active under oxidizing conditions. His-tagged SUMO protease (Ulp1), HRV 3C protease, and Enterokinase generally maintain activity. Avoid DTT or β-mercaptoethanol in your cleavage buffer.
- Protocol (SUMO cleavage): Purify the fusion protein via Ni-NTA under native conditions. Dialyze into cleavage buffer: 50 mM Tris-HCl (pH 8.0), 150 mM NaCl. Add SUMO protease (1:100 molar ratio) and incubate at 4°C for 16 hours or 25°C for 4 hours. Pass over Ni-NTA again to separate the cleaved tag (His-tagged) from your target protein.
Q4: What quantitative improvements can I expect from combining fusion tags and chaperone co-expression in disulfide-bond competent strains?
A: Performance varies, but typical outcomes are summarized below:
Table 1: Quantitative Outcomes of Combined Folding Strategies
| Strategy | Soluble Yield (mg/L culture) | % Active (Correctly Folded) | Key Metric vs. Baseline (BL21(DE3)) |
|---|---|---|---|
| Baseline (BL21(DE3) + Fusion Tag) | 2 - 10 | 10 - 30% | Reference |
| + Disulfide Strain (SHuffle) | 5 - 20 | 40 - 70% | 2-5x increase in active fraction |
| + DsbC Co-expression | 8 - 25 | 60 - 85% | Significant reduction in misfolded aggregates |
| + GroEL/GroES Co-expression | 10 - 30 | 20 - 50%* | 2-3x increase in soluble yield |
| Combined (SHuffle + Tag + DsbC + GroEL/ES) | 15 - 50+ | 70 - 95% | Maximizes both yield and correctness |
* Note: GroEL/ES primarily boosts solubility; activity gain depends on correct disulfide formation, which requires the oxidizing background.
Experimental Protocol: Integrated Co-expression for Disulfide Bond Formation
Title: Co-expression of Target Protein (MBP-Fusion), DsbC, and GroEL/GroES in SHuffle E. coli.
Materials:
- SHuffle T7 Competent E. coli cells.
- Plasmid 1 (pET-derived): Target gene cloned downstream of an MBP tag with a TEV protease site, Amp⁺.
- Plasmid 2 (pACYC-derived): Expressing dsbC gene, Cm⁺.
- Plasmid 3 (pGro7 or similar): Expressing groEL-groES operon, induced by L-arabinose, Spec⁺.
- LB media with appropriate antibiotics (Amp, Cm, Spec).
- 0.5 M IPTG, 20% (w/v) L-arabinose, 1 M Cysteine stock.
Method:
- Co-transformation: Transform plasmids 2 and 3 into SHuffle cells. Select on LB-Agar plates with Cm and Spec. Then, transform plasmid 1 into this strain and select on plates with Amp, Cm, and Spec.
- Inoculation & Growth: Pick a colony into 5 mL LB with all three antibiotics. Grow overnight at 30°C, 220 rpm.
- Main Culture: Dilute overnight culture 1:100 into fresh LB (+ antibiotics). Grow at 30°C to an OD₆₀₀ of 0.6.
- Chaperone Pre-induction: Add L-arabinose to a final concentration of 0.5 mg/mL. Continue incubation for 45-60 minutes.
- Target Induction: Add IPTG to a final concentration of 0.2 mM. Add filter-sterilized cysteine to 5 mM final concentration.
- Expression: Lower temperature to 25°C. Incubate with shaking for 16-20 hours.
- Harvest: Centrifuge cells at 4,000 x g for 20 min at 4°C. Process pellet for lysis and purification under native, non-reducing conditions.
Diagrams
Diagram 1: Strategy for Enhancing Disulfide Bond Folding in Cytoplasm
Diagram 2: Experimental Workflow for Co-expression
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Enhanced Cytoplasmic Folding Experiments
| Reagent / Material | Function & Rationale |
|---|---|
| SHuffle T7 Express E. coli (NEB) | Engineered strain with oxidized cytoplasm (ΔtrxB/gor) and periplasmic DsbC expressed cytoplasmically to catalyze disulfide bond isomerization. |
| pET MBP Fusion Vectors (e.g., pETM-41) | Provides a strong T7 promoter and Maltose-Binding Protein (MBP) tag to enhance solubility and provide an affinity handle. |
| pGro7 Plasmid (Takara Bio) | Chaperone plasmid expressing GroEL-GroES system; induced by L-arabinose to assist in folding and prevent aggregation. |
| pACYC184-dsbC Plasmid | Medium-copy plasmid compatible with ColE1 origins; used for constitutive or inducible expression of DsbC chaperone. |
| SUMO Protease (Ulp1) | Highly specific protease that cleaves after the SUMO tag; remains active under a wide range of conditions, including non-reducing buffers. |
| Ni-NTA Superflow Resin (Qiagen) | Immobilized metal-affinity chromatography resin for purifying His-tagged fusion proteins and His-tagged proteases. |
| Non-Reducing SDS-PAGE Buffer | Sample buffer lacking β-mercaptoethanol or DTT to allow assessment of disulfide bond formation via altered electrophoretic mobility. |
| L-Cysteine Hydrochloride | Added to culture media to support the redox pathway and provide a source of reducing equivalents for the Dsb system. |
Technical Support Center: Troubleshooting & FAQs
FAQs and Troubleshooting Guide
Q1: I am getting low scFv expression yields in the E. coli cytoplasm. What are the primary causes? A: Low yields are often due to codon bias, protein aggregation, or inefficient transcription/translation. Ensure you use an E. coli codon-optimized gene sequence. Switch to a weaker promoter (e.g., pTrc instead of T7) to reduce translation rate and aggregation. Optimize induction conditions (IPTG concentration, temperature, time).
Q2: My scFv is forming insoluble inclusion bodies. How can I improve soluble expression? A: This is common for proteins requiring disulfide bonds in the reducing cytoplasm. Implement the following:
- Use a mutated host strain: Use SHuffle T7 or Origami strains, which enhance cytoplasmic disulfide bond formation.
- Lower growth temperature: Induce at 18-25°C to slow folding and aggregation.
- Co-express chaperones: Use plasmids co-expressing GroEL-GroES or DnaK-DnaJ-GrpE.
- Optimize media: Test rich media (e.g., Terrific Broth) and additives like sorbitol, betaine, or ethanol.
Q3: How can I verify that intramolecular disulfide bonds are forming correctly in my cytoplasmic scFv? A: Perform a non-reducing vs. reducing SDS-PAGE analysis. A correctly folded scFv with disulfide bonds will migrate faster on non-reducing gels compared to the reduced, linear form. Confirm with mass spectrometry or functional assays (e.g., ELISA).
Q4: My purified scFv shows no antigen binding in ELISA. What could be wrong? A: Loss of function suggests misfolding. Ensure:
- The purification protocol (e.g., IMAC) includes a redox buffer to maintain disulfides.
- Refolding steps are necessary if purified from inclusion bodies. Use a redox couple (GSH/GSSG) during refolding.
- The linker between VH and VL domains (e.g., (G4S)3) is flexible and correctly encoded.
Q5: What are the key differences between periplasmic and cytoplasmic scFv production in the context of disulfide bond formation? A: See Table 1.
Table 1: Comparison of Cytoplasmic vs. Periplasmic scFv Production
| Parameter | Cytoplasmic Production (with Enhanced Strains) | Periplasmic Production |
|---|---|---|
| Oxidative Environment | Engineered to be oxidizing (e.g., trxB/gor mutations) | Naturally oxidizing |
| Disulfide Bond Formation | Facilitated by strains like SHuffle | Facilitated by Dsb enzymes |
| Yield | Typically higher total protein | Typically lower, but more soluble |
| Solubility | Can be low; requires optimization | Generally higher |
| Purification Complexity | Can be simpler (cell lysis) | Requires periplasmic extraction |
| Functional Folding Success | Variable, highly strain/method-dependent | Historically more reliable |
Detailed Experimental Protocols
Protocol 1: Cytoplasmic Expression of scFv in SHuffle T7 E. coli
- Cloning: Clone codon-optimized scFv gene into a cytoplasmic expression vector (e.g., pET series) with a solubility tag (e.g., MBP, Trx) if needed.
- Transformation: Transform chemically competent SHuffle T7 cells. Plate on LB agar with appropriate antibiotic (e.g., 100 µg/mL ampicillin).
- Culture & Induction: Inoculate 5 mL LB+antibiotic starter culture. Grow overnight at 30°C, 220 rpm. Dilute 1:100 into fresh medium. Grow at 30°C until OD600 ~0.6-0.8. Induce with 0.1-0.5 mM IPTG. Reduce temperature to 25°C. Incubate with shaking for 16-20 hours.
- Harvesting: Pellet cells at 4,000 x g for 20 min at 4°C. Store at -80°C or proceed.
Protocol 2: Analysis of Solubility and Disulfide Bond Formation
- Lysis: Resuspend pellet in Lysis Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mg/mL lysozyme, protease inhibitors). Incubate on ice 30 min. Sonicate on ice (10 pulses of 10 sec, 20% amplitude). Centrifuge at 16,000 x g for 30 min at 4°C.
- Solubility Check: Separate supernatant (soluble fraction) and pellet (insoluble fraction). Resuspend pellet in an equal volume of lysis buffer + 1% SDS. Analyze equal volumes of both fractions by SDS-PAGE.
- Disulfide Bond Check: Prepare two identical samples from the soluble fraction. Add 2X non-reducing sample buffer to one. Add 2X reducing buffer (with β-mercaptoethanol) to the other. Boil both for 5 min. Run on the same SDS-PAGE gel. A mobility shift indicates disulfide formation.
Protocol 3: IMAC Purification under Redox Conditions
- Buffer Preparation: Equilibration/Wash Buffer: 50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10-20 mM Imidazole, 1 mM GSH/GSSG (10:1 ratio). Elution Buffer: Same as above with 250-500 mM imidazole.
- Purification: Pass clarified lysate over pre-equilibrated Ni-NTA resin. Wash with 10 column volumes (CV) of Wash Buffer. Elute with 5 CV of Elution Buffer.
- Buffer Exchange: Use a PD-10 desalting column to exchange into final storage buffer (e.g., PBS). Analyze by SDS-PAGE and measure concentration.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Cytoplasmic scFv Production
| Item | Function & Rationale |
|---|---|
| SHuffle T7 E. coli cells | Engineered trxB/gor mutant with cytoplasmic DsbC for promoting correct disulfide bond formation. |
| pET Expression Vector | High-copy number vector with T7 promoter for tunable, strong expression. |
| Codon-Optimized scFv Gene | Maximizes translation efficiency in E. coli, preventing ribosomal stalling. |
| Redox Buffer (GSH/GSSG) | Maintains a redox equilibrium in purification buffers to prevent scrambling of formed disulfides. |
| Solubility Tag Plasmid (e.g., pMAL) | Vector for fusing scFv to tags like MBP to enhance solubility and folding. |
| Chaperone Co-expression Plasmid | Plasmid (e.g., pG-KJE8) expressing GroEL/GroES/DnaK to assist in proper protein folding. |
| Ni-NTA Resin | For Immobilized Metal Affinity Chromatography (IMAC) purification of His-tagged scFv. |
| Non-Reducing Sample Buffer | For SDS-PAGE analysis without breaking disulfide bonds to assess correct folding. |
Visualization Diagrams
Title: Cytoplasmic scFv Expression and Analysis Workflow
Title: Thesis Context: Solving scFv Misfolding in the Cytoplasm
Solving Production Hurdles: Troubleshooting Low Yield, Misfolding, and Inefficient Oxidation
Troubleshooting Guide & FAQs
FAQ 1: Why is my IAM (Iodoacetamide) alkylation assay showing high background alkylation even in reducing conditions, suggesting poor cysteine blocking?
- Answer: High background often indicates that the reducing agent (e.g., DTT, TCEP) was not fully removed or quenched before adding IAM. Residual reductant competes with IAM for free cysteines, leading to incomplete alkylation of reduced disulfides.
- Troubleshooting Protocol:
- Increase Alkylant Concentration & Time: After reduction, increase IAM concentration from the standard 10-15 mM to 50 mM and incubate in the dark at 25°C for 30-45 minutes.
- Desalt Efficiently: Use a robust desalting column (e.g., Zeba Spin Columns) or precipitation (TCA/Acetone) to completely remove reductants before alkylation. Perform buffer exchange into alkylation buffer (e.g., 50 mM Tris-HCl, pH 8.0) without thiols.
- Confirm Quenching: If using TCEP, ensure the alkylation pH is ≥7.0 for optimal reactivity. Verify the pH of your reaction mixture.
- Positive Control: Always run a fully reduced and alkylated control sample to establish the baseline for complete alkylation.
FAQ 2: My Mass Spectrometry analysis shows unexpected mass shifts. Are these due to disulfide scrambling or other artifacts?
- Answer: Unexpected mass shifts can arise from several sources. Disulfide scrambling (thiol-disulfide exchange) is a common culprit, especially during sample prep for MS under non-ideal pH conditions. Other causes include oxidation (Met, Trp), deamidation, or incomplete alkylation.
- Troubleshooting Protocol to Prevent Scrambling:
- Control pH During Digestion: Perform enzymatic digestion (e.g., with Trypsin) at a slightly acidic pH (~pH 6.0) to minimize thiol-disulfide exchange. Use buffers like ammonium acetate.
- Use Alkylating Agents Post-Digestion: For non-reducing MS analysis, alkylate free cysteines immediately after denaturation (with Guanidine HCl) and before digestion, using IAM or NEM (N-ethylmaleimide).
- Employ Quenching Agents: Add low molecular weight thiol scavengers like NEM or iodoacetic acid after digestion to alkylate any newly liberated thiols.
- Data Analysis: Use software (e.g., Byonic, Mascot) to search for cross-linked peptides (Cys-Cys) and specify variable modifications for both reduced/alkylated (carbamidomethyl, +57 Da) and oxidized (disulfide, -2 Da) cysteines.
FAQ 3: How do I distinguish between intramolecular and intermolecular disulfide bonds using non-reducing SDS-PAGE?
- Answer: Non-reducing SDS-PAGE (without β-mercaptoethanol/DTT) shows mobility shifts.
- Troubleshooting Protocol & Interpretation:
- Run Three Conditions: Load the same protein sample in three lanes: a) Native (non-denatured, non-reduced), b) Denatured but Non-Reduced (with SDS, no DTT), c) Fully Reduced (with SDS and DTT).
- Interpretation Table:
| Lane Condition | Intramolecular Disulfide Bond Present | Intermolecular Disulfide Bond Present |
|---|---|---|
| Non-Reduced (Denatured) | Faster migration than reduced form (compact structure). | Slower migration (higher oligomer band) than reduced monomer. |
| Fully Reduced | Slower migration (unfolded chain). | Monomer band only. |
| Key Diagnostic | Band shift between reduced and non-reduced lanes. | Presence of higher MW oligomer band in non-reduced lane that disappears in reduced lane. |
FAQ 4: In my LC-MS/MS data, I cannot confidently assign disulfide-linked peptide pairs. What step is critical for successful identification?
- Answer: The most critical step is generating high-quality MS/MS spectra of the cross-linked peptides. This often fails due to poor fragmentation or low abundance.
- Troubleshooting Protocol:
- Optimize Fragmentation: Use electron-transfer dissociation (ETD) or EThcD (ETD supplemented with HCD) in addition to standard HCD/CID. ETD preserves labile bonds like disulfides, providing clearer backbone fragmentation for linkage assignment.
- Enrichment Strategy: Use diagonal chromatography (2D non-reducing/reducing) or affinity purification to enrich for disulfide-linked peptides before MS injection.
- Software Search Parameters: Ensure your database search includes the correct mass difference for a disulfide bond (-2.016 Da for the bond, or the exact mass of your alkylating agent if reduced post-digestion). Manually validate software-assigned hits by inspecting spectra for key y/b or c/z ions from each peptide chain.
Table 1: Common Alkylating Reagents for Cysteine Modification in Disulfide Analysis
| Reagent | Target | Mass Addition (Da) | Key Property | Best Used For |
|---|---|---|---|---|
| Iodoacetamide (IAM) | Free Thiols (-SH) | +57.0215 | Irreversible alkylation. | Standard blocking of reduced cysteines in bottom-up proteomics. |
| N-Ethylmaleimide (NEM) | Free Thiols (-SH) | +125.0480 | Irreversible alkylation, faster than IAM. | Quick quenching of reductants; labeling at lower pH. |
| 4-Vinylpyridine | Free Thiols (-SH) | +105.0578 | Forms stable adduct. | Alternative to IAM, often used in peptide mapping. |
| Iodoacetic Acid | Free Thiols (-SH) | +58.0055 | Adds negative charge. | Shifts pI for electrophoresis-based assays. |
Table 2: Comparison of Mass Spectrometry Fragmentation Techniques for Disulfide Bond Analysis
| Technique | Principle | Pros for Disulfide Analysis | Cons |
|---|---|---|---|
| CID/HCD | Collision-induced dissociation; breaks weakest bonds. | Fast, sensitive, widely available. | Often cleaves disulfide bond itself before peptide backbone, losing linkage info. |
| ETD | Electron transfer induces backbone cleavage. | Preserves post-translational modifications (PTMs) like disulfides; excellent for longer, charged peptides. | Lower efficiency for low-charge-state or small peptides; slower. |
| EThcD | Hybrid of ETD and HCD. | Provides both ETD (preserves linkage) and HCD-type fragments; increases confidence in assignment. | More complex instrumentation and data analysis. |
Experimental Protocols
Protocol 1: IAM Alkylation Assay for Free Thiol Quantification Purpose: To quantify the number of free cysteines (reduced disulfides) in a protein sample relative to a fully reduced control. Reagents: Purified protein, Iodoacetamide (IAM), Dithiothreitol (DTT), Guanidine HCl, Tris-HCl buffer (pH 8.0), Zeba Spin Desalting Columns. Procedure:
- Denature & Reduce: Prepare two 100 µL aliquots of protein (1 mg/mL) in 6 M Guanidine HCl, 50 mM Tris-HCl, pH 8.0.
- Test Sample: Add no DTT.
- Fully Reduced Control: Add DTT to 10 mM final concentration.
- Incubate both at 37°C for 30 minutes.
- Desalt: Pass each sample through a separate Zeba column pre-equilibrated with 50 mM Tris-HCl, pH 8.0, to remove DTT (from control) and change buffer.
- Alkylate: To each sample, add IAM to a final concentration of 15 mM. Incubate in the dark at 25°C for 30 minutes.
- Quench: Add excess DTT (to 20 mM) to consume any remaining IAM.
- Analysis: Measure the UV absorbance at 280 nm and 250 nm. The increase in A250 (due to carbamidomethyl-cysteine formation) is proportional to the number of alkylated cysteines. Compare the test sample to the fully reduced control to calculate the percentage of free thiols/disulfide formation.
Protocol 2: Non-Reducing vs. Reducing SDS-PAGE for Disulfide Bond Typing Purpose: To visually assess the presence of intra- or intermolecular disulfide bonds. Reagents: Protein sample, 4X Non-Reducing Loading Buffer (no DTT/β-ME), 4X Reducing Loading Buffer (with DTT/β-ME), SDS-PAGE gel, Coomassie stain. Procedure:
- Prepare three samples for the same protein:
- A (Native): 5 µL protein + 5 µL water.
- B (Non-Reduced): 5 µL protein + 5 µL Non-Reducing Loading Buffer.
- C (Reduced): 5 µL protein + 5 µL Reducing Loading Buffer.
- Heat samples B and C at 95°C for 5 minutes. Do not heat sample A.
- Load all samples on an SDS-PAGE gel (10-20% gradient recommended).
- Run electrophoresis, stain with Coomassie Blue, and analyze banding patterns as per Table 1 in the FAQs.
Visualizations
Diagram Title: Workflow for Disulfide Bond Assessment in Recombinant Protein
Diagram Title: IAM Assay Logic: Interpreting Free Thiol Results
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Disulfide Bond Analysis |
|---|---|
| SHuffle T7 E. coli Strain | Engineered to have a more oxidizing cytoplasm and constitutively express disulfide bond isomerase (DsbC), enhancing correct disulfide formation in vivo. |
| Tris(2-carboxyethyl)phosphine (TCEP) | Strong, odorless, and stable reducing agent. Effective at a wider pH range than DTT. Used to fully reduce disulfide bonds for control experiments. |
| Iodoacetamide (IAM) | Alkylating agent that covalently modifies free thiol (-SH) groups, preventing re-oxidation and allowing for mass tagging during MS analysis. |
| N-Ethylmaleimide (NEM) | Fast-acting thiol alkylator used to quench reduction reactions or label cysteines at neutral to slightly acidic pH. |
| Guanidine Hydrochloride (GdnHCl) | Chaotropic denaturant. Unfolds proteins to expose all disulfide bonds and cysteines to solution reagents during reduction/alkylation steps. |
| Trypsin (Sequencing Grade) | Protease for digesting proteins into peptides for MS mapping. Must be used under controlled pH to minimize disulfide scrambling. |
| Zeba Spin Desalting Columns | Rapid, efficient spin columns for buffer exchange and removal of small molecules (like reductants) prior to alkylation or MS analysis. |
| Mass Spec-Compatible Surfactant (e.g., RapiGest) | Aids protein solubilization and digestion, but is easily cleaved and removed before LC-MS, preventing ion suppression. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My protein of interest is expressing but shows low solubility and activity. I suspect improper disulfide bond formation in the cytoplasm. What are the primary condition levers to adjust? A: For enhancing cytoplasmic disulfide bond formation in engineered E. coli strains (e.g., trxB/gor mutants, SHuffle), systematically optimize these three parameters:
- Temperature: Reduce from 37°C to 16-30°C. Lower temperatures slow protein synthesis, allowing better folding and disulfide formation.
- Inducer Concentration: Titrate IPTG from 10 µM to 1 mM. Use the lowest effective concentration to minimize aggregation.
- Media: Use rich media (e.g., TB) for yield or defined media for control. Supplement with redox additives like cystine (oxidized form) at 0.1-1 mM.
Q2: How do I determine the optimal induction temperature for my disulfide-bonded protein? A: Conduct a parallel expression test. Inoculate cultures in TB or defined media with appropriate antibiotics. Grow to mid-log phase (OD600 ~0.6-0.8), then induce with a standard IPTG concentration (e.g., 0.5 mM) and split into flasks incubated at 37°C, 30°C, 25°C, and 16°C. Harvest after 4-6 hours (37°C) or 16-20 hours (lower temps). Analyze solubility via SDS-PAGE of total vs. soluble fractions.
Q3: I am using a T7 system in SHuffle cells. High IPTG concentrations lead to inclusion bodies. What should I do? A: The T7 system is very strong. Perform an IPTG titration experiment. Induce cultures at a lower temperature (25°C) with varying IPTG concentrations. Compare yield and solubility.
Q4: What media additives specifically promote disulfide bond formation in the cytoplasm? A: Key additives include:
- Cystine: The oxidized dimer of cysteine, directly provides a disulfide source. Use at 0.1-1 mM.
- GSH/GSSG Buffers: Glutathione redox buffers can be added to fine-tune the cytoplasmic redox potential, though uptake is variable.
- Metal Ions: Ensure sufficient Cu²⁺ or Mn²⁺ is present in trace elements, as they can influence redox chemistry.
Q5: My protein expression yield is low after shifting to low-temperature induction. How can I improve it? A: Extend the post-induction time to 16-24 hours. Ensure adequate aeration during slow growth. Alternatively, use auto-induction media formulated for low-temperature expression, which can improve biomass and yield.
Table 1: Effect of Temperature and IPTG on Solubility of a Model Disulfide-Rich Protein (Single-Domain Antibody) in SHuffle T7 E. coli
| Induction Temperature (°C) | IPTG Concentration (mM) | Total Protein Yield (mg/L) | Soluble Fraction (%) | Relative Activity (%) |
|---|---|---|---|---|
| 37 | 0.5 | 45.2 | 15 | 10 |
| 30 | 0.5 | 38.7 | 55 | 65 |
| 30 | 0.1 | 32.1 | 80 | 85 |
| 25 | 0.1 | 25.5 | 90 | 95 |
| 16 | 0.05 | 18.8 | 95 | 98 |
Table 2: Impact of Media and Additives on Disulfide Bond Formation Efficiency
| Media Type | Additive (concentration) | Relative Oxidizing Power (CyDisSO assay) | Specific Yield (mg/g DCW) | Notes |
|---|---|---|---|---|
| LB | None | 1.0 (baseline) | 1.0 (baseline) | High growth, low solubility |
| Terrific Broth (TB) | None | 1.2 | 2.5 | Improved yield, potential redox variability |
| M9 Minimal | None | 0.8 | 0.7 | Reproducible redox, lower biomass |
| M9 Minimal | Cystine (0.5 mM) | 2.5 | 1.5 | Significantly enhances disulfide formation |
| TB | Cystine (0.5 mM) | 2.8 | 2.8 | Optimal combination for this model system |
Experimental Protocols
Protocol 1: IPTG & Temperature Optimization Screen
- Preparation: Transform plasmid into E. coli SHuffle T7 cells. Pick a single colony to inoculate 5 mL LB with antibiotics. Grow overnight at 30°C, 220 rpm.
- Inoculation: Dilute overnight culture 1:100 into 25 mL of TB media (+ antibiotics) in four 125 mL flasks. Grow at 30°C to OD600 ~0.6.
- Induction & Division: Add IPTG to each flask to final concentrations of 1.0 mM, 0.5 mM, 0.1 mM, and 0.05 mM. Immediately after adding IPTG, split each induced culture into four 50 mL tubes.
- Temperature Shift: Place the tubes into four separate shaking incubators set at 37°C, 30°C, 25°C, and 16°C.
- Harvest: Incubate for: 4-6h (37°C), 6-8h (30°C), 16h (25°C), 20-24h (16°C). Pellet cells by centrifugation at 4°C.
- Analysis: Resuspend pellets in lysis buffer, lyse by sonication. Centrifuge to separate soluble and insoluble fractions. Analyze by SDS-PAGE (with/without DTT) and Western blot.
Protocol 2: Media Supplementation with Cystine
- Media Prep: Prepare standard TB and M9 media. Autoclave. Prepare a sterile-filtered 100 mM cystine stock solution in 0.1 M HCl (requires gentle heating to dissolve).
- Supplementation: Aseptically add cystine stock to cooled media to final concentrations of 0.1 mM, 0.5 mM, and 1.0 mM. Include a no-additive control.
- Expression: Inoculate and grow cultures as in Protocol 1. Induce all cultures with the predetermined optimal IPTG concentration and temperature.
- Assessment: Harvest cells. Measure final OD600 and wet cell weight. Process for solubility analysis. Quantify active protein using an activity assay (e.g., ELISA) relative to a purified standard.
Diagrams
Title: Optimization Workflow for Redox Control
Title: Parameters for Cytoplasmic Disulfide Bonds
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| SHuffle T7 E. coli Cells | Engineered to have an oxidizing cytoplasm (ΔtrxB/gor) and express disulfide bond isomerase (DsbC) in the cytoplasm to facilitate correct pairing. |
| pET Expression Vectors | High-copy number plasmids with T7 promoter for strong, inducible expression of the target protein. |
| Terrific Broth (TB) Media | Rich media providing high biomass yield, often leading to higher total protein expression levels. |
| M9 Minimal Media | Defined chemical composition allows precise control over redox additives and metal ions, improving reproducibility. |
| Cystine (Oxidized) | Disulfide-bonded dimer of cysteine. Added to media to provide an external source of disulfides, shifting the cytoplasmic redox potential. |
| Isopropyl β-d-1-thiogalactopyranoside (IPTG) | Non-metabolizable inducer for the lac and T7 promoter systems. Concentration is critical for tuning expression rate. |
| CyDisSO Dye (e.g., from Cytiva) | Cell-permeable fluorescent probe that reacts with the reducing environment. Fluorescence decreases as the cytoplasm becomes more oxidizing, allowing quantification of redox state. |
| BugBuster or Lysozyme/Lysis Reagents | For gentle cell lysis to preserve soluble protein and avoid artifactual oxidation or reduction during extraction. |
| Non-Reducing SDS-PAGE Sample Buffer | Contains SDS but no DTT/β-mercaptoethanol, allowing visualization of disulfide-bonded protein species by gel shift. |
Technical Support Center: Troubleshooting Guide & FAQs for Enhancing Disulfide Bond Formation in E. coli Cytoplasm
This support center provides targeted solutions for common experimental challenges in producing disulfide-bonded proteins in the E. coli cytoplasm, a key focus of ongoing thesis research on redox pathway engineering.
Frequently Asked Questions (FAQs)
Q1: My target protein with multiple disulfide bonds is entirely insoluble in the E. coli cytoplasm, even when using SHuffle strains. What should I try first? A: First, verify the redox state of your host. SHuffle strains constitutively express the trxB and gor mutations, but ensure you are using the correct antibiotic selection. Next, co-express a chaperone pair like GroEL/GroES or DnaK/DnaJ/GrpE to assist with folding. Consider N-terminal fusion tags such as Mxe GyrA intein or SUMO, which are highly soluble and can be cleaved off post-purification.
Q2: I see significant protein aggregation despite solubility enhancers. How can I optimize my fed-batch conditions to mitigate this? A: Aggregation often correlates with high specific production rates. In fed-batch, implement an exponential feeding strategy that limits the specific growth rate (μ) during the induction phase. Keep μ below 0.15 h⁻¹ to reduce metabolic burden and the rate of protein synthesis, allowing folding machinery (chaperones, DsbC) to keep pace. Simultaneously, lower the induction temperature to 20-25°C.
Q3: How do I choose between co-expressing cytoplasmic DsbC and molecular chaperones? A: Their functions are complementary. Use the decision logic below:
- If your protein has known slow-folding intermediates prone to misfolding, prioritize chaperones (GroEL/S or DnaK/J).
- If your protein requires concurrent formation of multiple disulfides that may form incorrectly, prioritize cytoplasmic DsbC for its isomerase activity.
- For complex proteins, use both systems simultaneously. Co-transform compatible plasmids (e.g., pGro7 for GroEL/S and pBad-DsbC vectors with different origins of replication and antibiotic resistance).
Q4: What are the key monitoring points in a fed-batch process for this application? A: Beyond standard OD₆₀₀ and substrate feed, monitor:
- Dissolved Oxygen (DO): Spikes indicate feeding imbalance or metabolic stress.
- Culture Redox Potential (ORP): A shift towards more oxidizing conditions post-induction can correlate with disulfide bond formation.
- Acetate Accumulation: Keep < 2 g/L via controlled feeding; acetate stresses the folding environment.
- Sample for Solubility: Take samples pre- and post-induction, lysing cells under non-denaturing conditions for SDS-PAGE analysis of soluble vs. insoluble fractions.
Experimental Protocols
Protocol 1: Screening for Optimal Solubility Enhancers Objective: Identify the best chaperone/fusion tag combination for your target protein.
- Clone Generation: Clone your target gene into a set of compatible E. coli expression vectors (e.g., pET series) featuring different N-terminal solubility tags (His-SUMO, His-MBP, His-Trx) and a control with only a His-tag.
- Co-transformation: Transform each construct into your base strain (e.g., SHuffle T7 Express) alongside a suite of chaperone plasmids (e.g., pGro7, pKJE7, pTf16). Include a "chaperone-only" control.
- Micro-scale Expression: Inoculate 2 mL deep-well blocks. Grow at 30°C to mid-log phase (OD₆₀₀ ~0.6). Induce with appropriate agent (IPTG for T7, arabinose for pBad). Reduce temperature to 20°C. Shake for 18-24 hours.
- Solubility Analysis: Pellet cells. Lyse using chemical (BugBuster) or enzymatic (lysozyme) method. Centrifuge at 15,000 x g for 20 min. Separate supernatant (soluble) and resuspended pellet (insoluble) fractions. Analyze by SDS-PAGE and Western blot.
Protocol 2: Fed-Batch Process Development for Aggregation Control Objective: Establish a reproducible fed-batch process minimizing aggregation.
- Seed Train: Prepare a primary seed in a shake flask from a glycerol stock. Inoculate a secondary seed in a larger flask to achieve sufficient biomass.
- Bioreactor Batch Phase: Inoculate bioreactor to initial OD₆₀₀ of ~0.1. Allow cells to grow in defined rich medium until the carbon source (e.g., glycerol) is nearly depleted, indicated by a sharp DO rise.
- Fed-Batch Induction Phase:
- Initiate an exponential feed of feed medium (e.g., 500 g/L glycerol, 10 g/L MgSO₄) to maintain a low, constant growth rate (μ = 0.10-0.12 h⁻¹).
- Simultaneously, induce protein expression with a low concentration of inducer (e.g., 0.1 mM IPTG).
- Maintain temperature at 25°C, pH at 6.8-7.2, and DO >30% via cascade control.
- Harvest: 6-8 hours post-induction, rapidly cool the culture and harvest by centrifugation. Process immediately or freeze at -80°C.
Data Presentation
Table 1: Comparison of Solubility Enhancement Strategies for Disulfide-Bonded Proteins in E. coli Cytoplasm
| Strategy | Example Agents/Genes | Primary Mechanism | Typical Yield Improvement* | Key Consideration |
|---|---|---|---|---|
| Chaperone Co-expression | GroEL/GroES, DnaK/DnaJ/GrpE | Prevent misfolding, provide folding chamber | 2- to 10-fold soluble yield | ATP-dependent; may burden cell. |
| Fusion Tags | MBP, SUMO, NusA, Trx | Increase intrinsic solubility, improve translation | 5- to 50-fold soluble yield | Requires cleavage step; tag can influence activity. |
| Cytoplasmic Disulfide Isomerase | DsbC (catalytically active in cytosol) | Catalyzes disulfide shuffling/isomerization | 3- to 20-fold active yield | Requires thioredoxin/glutaredoxin knockout background. |
| Fed-Batch Process Control | Exponential feed, low μ, low T | Reduces metabolic burden, slows synthesis | Varies; can double titer vs. batch | Complex setup; requires optimization for each strain/protein. |
*Improvement is highly protein-dependent and relative to baseline in standard SHuffle strain under batch conditions.
Table 2: Fed-Batch Parameters for Aggregation Minimization
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| Induction Temperature | 20-25°C | Slows protein synthesis, favors proper folding. |
| Post-Induction Specific Growth Rate (μ) | 0.10 - 0.15 h⁻¹ | Balances protein production with cellular folding capacity. |
| Inducer Concentration | Reduced (e.g., 0.05-0.2 mM IPTG) | Limits transcription/translation rate, reducing misfolding load. |
| Dissolved Oxygen | >30% | Prevents anaerobic stress and improper oxidation. |
| Acetate Concentration | < 2 g/L | High acetate inhibits growth and disrupts protein folding. |
Visualizations
Diagram 1: Decision Pathway for Mitigating Aggregation
Diagram 2: Fed-Batch Workflow for Folding Optimization
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| SHuffle T7 Express E. coli | Engineered strain with cytoplasmic DsbC and trxB/gor deletions for disulfide bond formation. |
| pGro7 Plasmid | Chaperone plasmid expressing GroEL/GroES; chloramphenicol resistant. |
| pBad-DsbC (cytoplasmic) Plasmid | Plasmid for arabinose-induced expression of DsbC targeted to the cytoplasm. |
| BugBuster Master Mix | Gentle, non-denaturing detergent for cell lysis and soluble protein extraction. |
| His-Tagged Fusion Vectors (pET-SUMO, pET-MBP) | Expression vectors for testing solubility-enhancing N-terminal fusion tags. |
| TEV or SUMO Protease | For cleaving off solubility fusion tags after purification. |
| Defined Medium (e.g., M9 minimal salts + glucose/glycerol) | Essential for reproducible fed-batch process development and metabolic control. |
| Anti-DsbC Antibody | For verifying DsbC expression and stability via Western blot. |
| Redox Sensor (e.g., roGFP2-Orp1) | Genetically encoded probe to monitor real-time cytoplasmic redox state. |
Technical Support Center: Troubleshooting Guide & FAQs
FAQs & Troubleshooting for Enhanced Disulfide Bond Formation in E. coli Cytoplasm
Q1: My target protein with multiple disulfide bonds is expressed but remains entirely in the insoluble fraction, even when using oxidative cytoplasm strains (e.g., SHuffle). What should I check? A: This is a classic issue of imbalanced oxidation and folding. Investigate the following:
- Expression Rate: Your target protein may be expressed too rapidly, overwhelming the catalytic folding machinery. Solution: Reduce expression temperature (e.g., to 20-25°C), use a lower inducer concentration (e.g., 0.05-0.1 mM IPTG), or switch to an auto-induction medium.
- Catalyst-to-Target Ratio: The expression level of disulfide bond catalysts (DsbC, DsbG) may be insufficient relative to your target. Solution: Use a plasmid system where catalysts are expressed from a stronger promoter than the target protein, or pre-induce catalyst expression 1-2 hours before inducing target protein expression.
- Solubility Tag: Consider using a fusion partner like maltose-binding protein (MBP) or thioredoxin (Trx) to enhance initial solubility and provide more time for oxidation.
Q2: How do I choose between strains like SHuffle T7, Origami 2, and Rosetta-gami 2? A: The choice depends on your target protein's requirements.
| Strain | Key Genotype | Primary Use Case | Notes |
|---|---|---|---|
| SHuffle T7 | Δgor ΔtrxB dsbC (cytoplasmic), degP |
Strongest oxidative power. For difficult, multiple disulfide bond proteins. | DsbC is constitutively expressed in the cytoplasm. T7 RNA polymerase allows high-level expression. |
| Origami 2 | Δgor ΔtrxB |
General cytoplasmic oxidation. Provides a stabilized oxidizing environment. | Lacks specific catalysts; relies on host glutaredoxins/thioredoxins. Weaker than SHuffle. |
| Rosetta-gami 2 | Δgor ΔtrxB + Rosetta tRNA genes |
Oxidation for proteins with rare codons. Combines oxidation power with enhanced translation. | Useful if your gene has codons rarely used in E. coli (e.g., Arg: AGA, AGG; Pro: CCC). |
Q3: Expression is fine, but my purified protein shows heterogeneous disulfide bonding or is inactive. How can I improve fidelity? A: This suggests incorrect disulfide pairing (misfolding).
- Co-express a chaperone: Co-express cytoplasmic chaperones like GroEL/ES or DnaK/DnaJ/GrpE to prevent aggregation and provide more opportunities for correct folding. Use a compatible plasmid with a different origin of replication and antibiotic resistance.
- Optimize redox buffer: During lysis and purification, include a redox buffer (e.g., 1-5 mM reduced/oxidized glutathione or cysteine/cystine) to maintain existing disulfides and prevent scrambling.
- Screen expression conditions: Perform a matrix screen varying induction OD, temperature, and induction length. Analyze both soluble yield and activity.
Q4: What are the key reagents and controls for monitoring the success of cytoplasmic disulfide bond formation? A:
Research Reagent Solutions
| Reagent | Function | Example/Catalog # |
|---|---|---|
| AMS (4-Acetamido-4'-maleimidylstilbene-2,2'-disulfonic acid) | Thiol alkylating agent. Adds ~500 Da per free cysteine, causing a gel shift. Essential for assessing oxidation state. | Thermo Fisher, A485 |
| DTNB (Ellman's Reagent) | Quantify free thiols in purified protein samples. | Sigma Aldrich, D8130 |
| PNGase F | Deglycosylase. Rule out glycosylation if activity is low (ensure protein is not modified in unexpected ways). | NEB, P0704 |
| Reducing vs. Non-Reducing SDS-PAGE | Fundamental assay. Compare migration with and without β-mercaptoethanol/DTT to detect disulfide-stabilized complexes or compact folds. | - |
| Glutathione Redox Buffers | Maintain redox potential in purification buffers to prevent post-lysis scrambling. | Sigma Aldrich, G6529 (Oxidized), G6529 (Reduced) |
Experimental Protocol: Assessing Disulfide Bond Formation via AMS Alkylation Objective: Determine if cysteine residues in your target protein are oxidized (forming disulfides) or reduced (free thiols).
- Harvest & Lysis: Induce expression, harvest cells, and lyse using a non-reducing method (e.g., BugBuster without DTT, or lysozyme/freeze-thaw).
- Alkylation Reaction: Split lysate into two 50 µL aliquots.
- Sample +AMS: Add AMS to 15 mM final concentration. Incubate in dark, 30 min, RT.
- Control: Add equivalent volume of water.
- Analysis: Run both samples on non-reducing SDS-PAGE (do not boil with reducing agent!). A significant upward shift for the +AMS sample indicates the presence of free thiols (reduced cysteines). No shift suggests cysteines are already oxidized (in disulfides) and protected from alkylation.
Experimental Protocol: Co-expression of Target Protein and Chaperone Objective: Enhance correct folding by providing chaperone assistance.
- Strain: Use your chosen oxidative strain (e.g., SHuffle T7).
- Plasmids:
- Plasmid 1: Target gene in a vector with one origin of replication (e.g., ColE1) and antibiotic resistance (e.g., AmpR).
- Plasmid 2: Chaperone genes (e.g., groEL/ES operon) in a compatible vector with a different origin (e.g., p15A) and resistance (e.g., CmR).
- Transformation: Co-transform both plasmids or transform the chaperone plasmid first, then the target plasmid.
- Expression: Grow culture with both antibiotics. Induce chaperone expression first (if under inducible control, e.g., with arabinose), then induce target protein expression later (e.g., with IPTG). Proceed to purification.
Visualizations
Title: Workflow for Cytoplasmic Disulfide Bond Expression Optimization
Title: Disulfide Bond Formation and Isomerization Pathway
Technical Support Center
Troubleshooting Guide
Problem 1: Reduced Target Protein Yield and Increased Aggregation in Bioreactor vs. Shake Flask
- Symptoms: High expression in shake flasks, but low soluble yield and high inclusion body formation in the bioreactor.
- Likely Cause: Inefficient disulfide bond formation due to a more reduced cytoplasm in the controlled bioreactor environment, exacerbated by higher cell densities and different mixing/aeration.
- Solution: Implement a real-time Redox Potential (ORP) monitoring probe. Consider adding controlled, low amounts of oxidizing agents (e.g., Cu²⁺ phenanthroline) or redox-mediating supplements (e.g., cystine) to the fed-batch media. Optimize the dissolved oxygen (DO) setpoint and agitation strategy to influence the intracellular redox state.
Problem 2: Inconsistent Disulfide Bond Formation Between Batches
- Symptoms: Variable product quality (oxidized vs. reduced species) despite identical genetic constructs and media recipes.
- Likely Cause: Differences in the metabolic state and NADPH/NADP+ ratio at induction between scales, affecting the thioredoxin and glutathione pathways.
- Solution: Standardize the pre-induction growth phase by inducing at a consistent physiological state (e.g., specific glucose consumption rate, not just OD). Use a defined feed strategy to control growth rate and metabolic byproduct accumulation.
Problem 3: Cell Viability Drop Post-Induction in Bioreactor
- Symptoms: Rapid decline in viability following induction of disulfide-bonded protein, not observed in flasks.
- Likely Cause: Metabolic burden and redox stress from futile cycling of incorrect disulfide bonds, depleting reducing equivalents (NADPH) and generating reactive oxygen species (ROS).
- Solution: Lower the induction temperature (e.g., to 25-30°C). Reduce the inducer concentration (e.g., IPTG). Co-express chaperones (e.g., DsbC) or redox regulators (e.g., gor, trxB mutants) to alleviate stress.
Frequently Asked Questions (FAQs)
Q1: Why is redox balance more challenging to maintain in a bioreactor than in a shake flask? A: Shake flasks have limited control and gradients (O₂, pH, nutrients). Bioreactors provide homogeneous, tightly controlled conditions that can lead to different metabolic fluxes. High cell densities in bioreactors increase oxygen demand and byproduct accumulation (e.g., acetate), which can alter the NADH/NAD+ and NADPH/NADP+ pools, directly impacting cytoplasmic redox homeostasis and disulfide bond formation pathways.
Q2: What key parameters should I monitor when scaling up my E. coli disulfide bond formation experiment? A: Beyond standard pH, DO, and temperature, monitor:
- Redox Potential (ORP): Direct indicator of the broth's oxidative state.
- Respiratory Quotient (RQ): Indicates metabolic shift between respiration and fermentation.
- Specific Consumption/Rates: For glucose, oxygen, and alkali (for pH control). Sudden changes can signal redox stress.
- Off-gas analysis (O₂, CO₂): For real-time metabolic insight.
Q3: Are there specific E. coli strains or genetic constructs recommended for bioreactor scale-up of cytoplasmic disulfide bond formation? A: Yes. The most robust systems combine:
- Strain: E. coli SHuffle T7 or Origami 2 with mutations in the thioredoxin (trxB) and glutathione reductase (gor) pathways to create a more oxidizing cytoplasm.
- Plasmid: Use a vector with a strong, tunable promoter (e.g., pET with T7/lac). For complex proteins, include a fusion tag that enhances solubility (e.g., MBP, SUMO) and a signal for translocation to the more oxidizing periplasm if needed.
Q4: How can I adjust my feeding strategy to support redox balance? A: Avoid glucose-only feeds that can cause overflow metabolism and acetate production. Use:
- Mixed Substrate Feeding: Combine glucose with a less repressing carbon source (e.g., glycerol, fructose) at a low, constant rate.
- Exponential Feeding: Match the feed rate to the desired specific growth rate (μ) to prevent nutrient limitation or excess.
Data Presentation
Table 1: Comparison of Key Parameters Affecting Redox Balance: Shake Flask vs. Bioreactor
| Parameter | Typical Shake Flask Condition | Controlled Bioreactor Condition | Impact on Cytoplasmic Redox & Disulfide Bonds |
|---|---|---|---|
| Dissolved O₂ | Fluctuating, often low (<20% saturation) | Precisely controlled (e.g., 30-40% saturation) | High, stable O₂ can increase ROS, stress pathways, and potentially aid oxidation. |
| pH | Uncontrolled, declines with growth | Tightly controlled (e.g., pH 7.0) | Optimal pH stability supports enzyme function (e.g., Dsb enzymes, isomerases). |
| Mixing / Shear | Low, uneven mixing | High, homogeneous mixing | Better mixing improves nutrient/O₂ distribution but can cause local shear stress. |
| Cell Density | Limited (OD~5-15) | High (OD 50-100+) | High density leads to nutrient gradients, byproduct accumulation, and metabolic stress. |
| Substrate Availability | Batch, declining concentration | Fed-batch, controlled delivery | Prevents catabolite repression and acetate formation, stabilizing metabolism. |
| Redox Potential (ORP) | Not measured, variable | Can be monitored and logged | Direct correlation with the oxidative capacity of the environment. |
| Heat Transfer | Limited, follows incubator | Actively controlled | Precise temperature control post-induction is critical for folding. |
Table 2: Research Reagent Solutions for Enhanced Cytoplasmic Disulfide Bond Formation
| Reagent / Material | Function in Experiment | Key Consideration for Scale-Up |
|---|---|---|
| SHuffle T7 Express E. coli | Strain with mutated trxB & gor pathways and cytoplasmic DsbC for disulfide bond formation. | Ensure genetic stability over long bioreactor runs; monitor plasmid retention. |
| pET-based Expression Vector | Provides strong, IPTG-inducible T7 promoter for target protein expression. | Lower IPTG concentrations (0.05-0.2 mM) often sufficient in high-density culture. |
| Terrific Broth (TB) / Defined Media | Rich or defined media supporting high cell density. | Defined media (e.g., M9+feed) improves reproducibility and simplifies downstream. |
| Redox Mediator: Cystine | Oxidized form of cysteine; can be imported and contribute to the cellular redox buffer. | Add in small, controlled amounts (0.1-1 mM) to feed medium to avoid toxicity. |
| Chaperone Plasmid (e.g., pG-KJE8) | Co-expresses GroEL/ES and DnaK/DnaJ/GrpE chaperone systems. | Adds metabolic burden; optimize inducer (arabinose, tetracycline) concentration. |
| Metal Cofactor: Cu²⁺-1,10-phenanthroline | Artificial oxidant that can catalyze disulfide formation in the cytoplasm. | Use at very low concentrations (µM range); can be toxic and non-specific. |
| Feed Solution: Glycerol/Glucose Mix | Carbon source for fed-batch fermentation. | Mixed feeds can reduce acetate and support better redox management than glucose alone. |
Experimental Protocols
Protocol 1: Scaling Up Expression from Flask to Bioreactor
- Inoculum Train: Start from a single colony in LB + antibiotic. Grow a 5 mL overnight culture (16-18 hrs, 30°C, 250 rpm). Dilute 1:100 into 500 mL shake flask with main culture media. Grow to mid-log phase (OD600 ~0.6-0.8). This is the bioreactor inoculum.
- Bioreactor Setup (3L vessel, 1.5L working volume): Calibrate pH and DO probes. Sterilize in-situ with base media. Set initial conditions: Temperature = 30°C, pH = 7.0 (controlled with 25% NH₄OH and 15% H₃PO₄), Airflow = 1.0 vvm, Agitation = 500-800 rpm, DO = 40% (cascaded to agitation and O₂ enrichment).
- Batch Phase: Transfer inoculum to achieve starting OD600 ~0.1. Allow cells to consume batch carbon source (e.g., 10 g/L glycerol). Monitor DO spike indicating batch depletion.
- Fed-Batch Phase: Initiate exponential feed of carbon source (e.g., 500 g/L glycerol solution) to maintain a desired specific growth rate (μ = 0.15 h⁻¹). Continue until target cell density (OD600 ~50) is reached.
- Induction: Lower temperature to 25°C. Add IPTG to a final, optimized concentration (e.g., 0.1 mM). Optionally, add redox mediator (e.g., 0.5 mM cystine) to the feed.
- Post-Induction: Maintain conditions for 4-16 hours. Monitor viability (via stains), ORP, and RQ.
- Harvest: Chill culture to 4°C and harvest via continuous-flow centrifugation.
Protocol 2: Analyzing Disulfide Bond Formation & Redox State
- Sample Collection: Rapidly withdraw culture samples pre- and post-induction. Immediately plunge into dry ice/ethanol to quench metabolism.
- Intracellular Redox State (Glutathione): Thaw sample in 0.1% TCA. Derivatize with monobromobimane. Analyze reduced (GSH) and oxidized (GSSG) glutathione via HPLC with fluorescence detection. Calculate GSH/GSSG ratio.
- Protein Solubility & Oxidation: Lyse cells via sonication in non-reducing buffer (+ protease inhibitors, NEM to block free thiols). Separate soluble and insoluble fractions by centrifugation. Analyze both fractions by non-reducing SDS-PAGE (no β-mercaptoethanol).
- Mass Confirmation: Perform LC-MS on the purified target protein to confirm the mass of the correctly oxidized species versus the reduced form.
Visualizations
Diagram Title: Scale-Up Challenges Impacting Disulfide Bond Formation
Diagram Title: Optimized Bioreactor Workflow for Redox Control
Diagram Title: Strategies & Analytics for Redox Balance
Benchmarking Success: How to Validate Disulfide Bonds and Compare Leading E. coli Strain Platforms
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My LC-MS/MS analysis of tryptic peptides shows no ions corresponding to disulfide-linked peptides. What could be wrong? A: This is often due to insufficient reduction of disulfide bonds prior to digestion or incorrect digestion conditions.
- Solution 1: Verify your reduction protocol. Use 5-10 mM DTT or TCEP at 56°C for 30-45 minutes. For proteins expressed in the E. coli cytoplasm (a reducing environment), ensure you first fully oxidize/denature the protein to expose all bonds.
- Solution 2: Check enzyme specificity. Use sequencing-grade trypsin and ensure digestion pH is 7.5-8.5. Denaturation in 6 M guanidine-HCl or 8 M urea prior to reduction may be necessary.
- Solution 3: Confirm MS acquisition method. Use positive ion mode and a data-dependent MS2 method with collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD). Set an appropriate m/z range (e.g., 300-2000).
Q2: I see a mass shift in my intact protein analysis under non-reducing conditions, but cannot pinpoint the disulfide bonds. A: This indicates successful formation but requires mapping.
- Solution: Employ a middle-down or top-down MS approach. Use enzymes like Glu-C or Asp-N under non-reducing conditions to generate larger fragments. Perform ETD or EThcD fragmentation alongside HCD, as these methods better preserve labile modifications like disulfides for direct localization.
Q3: My data is noisy, and I cannot confidently assign disulfide-linked peptide spectra. A: This is common with complex backgrounds or partial oxidation.
- Solution 1: Improve chromatographic separation. Use a longer C18 column (e.g., 150-250 mm), a shallower gradient (e.g., 0.5% B/min), and a lower flow rate (e.g., 0.3 µL/min for nanoLC).
- Solution 2: Utilize data processing software features. Use search engines like Mascot, ProteinPilot, or Byonic with "cross-link" search settings. Manually validate hits by checking for characteristic fragment ions (e.g., neutral losses of 64 Da [SO2] from cysteine sulfenic acid).
Q4: How do I distinguish between native disulfides and non-native, scrambled bonds in my E. coli cytoplasm expression system? A: This is a critical validation step for the thesis context.
- Solution: Perform a time-course analysis. Quench folding/oxidation at different time points (seconds to minutes) using acid (e.g., 1% TFA). Map disulfides at each point. Native bonds form faster and more consistently. Compare to a known standard or the native protein pattern.
Q5: My protein contains multiple cysteines. How do I map all possible disulfide pairings? A: Use a combination of enzymatic digests.
- Solution: Create a parallel digestion strategy. Digest the non-reduced protein with two different enzymes (e.g., trypsin and chymotrypsin). This generates overlapping peptide fragments, increasing the chance of isolating each disulfide bond within a measurable peptide. Combine the results for complete mapping.
Detailed Experimental Protocol: Disulfide Mapping via LC-MS/MS
Objective: To identify and confirm specific disulfide bond linkages in a recombinant protein expressed in the E. coli cytoplasm.
Materials:
- Purified protein sample (> 90% purity)
- Digestion Buffer: 50 mM Tris-HCl, pH 8.0, 1 M Guanidine-HCl
- Reducing Agent: 100 mM Dithiothreitol (DTT) stock
- Alkylating Agent: 300 mM Iodoacetamide (IAM) stock
- Quenching Solution: 100 mM DTT
- Enzymes: Sequencing-grade trypsin, Glu-C
- Solvents: 0.1% Formic Acid (FA) in water (Solvent A), 0.1% FA in 80% Acetonitrile (Solvent B)
- LC-MS/MS System: Nanoflow UHPLC coupled to a Q-Exactive HF or Orbitrap Fusion mass spectrometer.
Procedure:
- Denaturation & Alkylation (for reduced control): Dilute 20 µg protein in 20 µL digestion buffer. Add DTT to 5 mM, incubate 30 min at 56°C. Cool, add IAM to 15 mM, incubate 20 min in the dark. Quench with excess DTT.
- Non-Reduced Sample Prep: Dilute 20 µg protein in 20 µL 50 mM Tris-HCl, pH 8.0. Do not add DTT or IAM.
- Digestion: For both samples, add trypsin at a 1:20 (w/w) enzyme-to-protein ratio. Incubate 4-16 hours at 37°C.
- LC-MS/MS Analysis:
- Column: 75 µm x 250 mm, C18 (1.6 µm beads).
- Gradient: 2% B to 35% B over 90 min, then to 95% B in 5 min.
- MS1: Resolution 120,000, scan range 350-1600 m/z.
- MS2: Top 20 precursors, HCD fragmentation at 28% normalized collision energy, resolution 15,000.
- Data Analysis: Search reduced sample data against protein sequence to confirm coverage. Search non-reduced data using a specialized disulfide search algorithm (e.g., Mascot's "Disulfide Bridge" feature or Byonic's "Common Biological Modifications" set to include disulfide).
Table 1: Common Disulfide Mapping Mass Spectrometry Parameters & Outcomes
| Parameter | Typical Value/Range | Purpose/Impact |
|---|---|---|
| MS1 Resolution | 60,000 - 120,000 | Accurate parent ion mass determination; distinguishes charge states. |
| HCD NCE | 25-32% | Optimized for peptide backbone cleavage while observing disulfide-specific fragments. |
| Chromatographic Gradient | 60-120 min | Resolves complex peptide mixtures; critical for separating linked peptides. |
| Expected Mass Shift (Non-Reduced vs. Reduced) | -2 Da per disulfide bond (loss of 2 H) | Intact mass check confirms total number of bonds formed. |
| Confidence Threshold (Peptide Spectral Match, PSM) | ≤ 1% FDR | Standard for high-confidence identification of disulfide-linked peptides. |
Table 2: Troubleshooting Common LC-MS/MS Issues for Disulfide Mapping
| Observed Problem | Potential Cause | Recommended Action |
|---|---|---|
| Low signal for disulfide peptides | Poor ionization efficiency; co-elution | Add post-column 50% IPA infusion to boost signal; optimize LC gradient. |
| Inconsistent bond identification | Partial reduction in sample | Include alkylation step without prior reduction for non-reduced sample. |
| High spectral complexity | Non-specific cleavage | Use higher grade enzyme, check digestion pH and time, purify protein better. |
| Software cannot assign bonds | Unusual cysteine spacing | Use complementary enzyme (Glu-C), consider ETD fragmentation. |
Diagrams
Title: Disulfide Mapping MS Workflow for E. coli Cytoplasm Proteins
Title: Decision Tree for Disulfide MS Data Interpretation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Disulfide Mapping & Validation
| Item | Function in Context of E. coli Cytoplasm Research |
|---|---|
| Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agent; more stable than DTT, effective at low pH, used to quench reactions or create reduced controls. |
| Guanidine Hydrochloride (Gdn-HCl) | Chaotropic denaturant. Unfolds proteins from E. coli lysates, exposing all cysteines for analysis or controlled re-oxidation. |
| Iodoacetamide (IAM) | Alkylates free thiols (-SH). Used to "cap" reduced cysteines after reduction, preventing reformation or scrambling of disulfides. |
| Sequencing-Grade Trypsin | Protease for bottom-up MS. High purity minimizes non-specific cleavage, ensuring reliable peptide maps for disulfide assignment. |
| C18 StageTips / Spin Columns | For sample cleanup and desalting of peptides prior to LC-MS. Removes salts and detergents that interfere with ionization. |
| Cystine/Cysteine Redox Buffer | Defined redox couple (e.g., GSH/GSSG). Used in in vitro refolding experiments to mimic the periplasmic environment and promote correct disulfide bonding. |
| High-pH Reverse-Phase LC Columns | For 2D-LC separations. Fractionates complex peptide mixtures by charge/hydrophobicity in first dimension, increasing depth of analysis for hard-to-detect links. |
| ETD/ECD-Compatible Mass Spectrometer | Enables electron-driven fragmentation (ETD, EThcD) which preserves labile disulfide bonds, providing direct sequence and linkage information. |
Troubleshooting Guide & FAQs
Q1: In my ELISA for a cytoplasmically expressed disulfide-bonded protein, I get high background noise. What could be the cause? A: High background in ELISA for recombinant E. coli cytoplasmic proteins is often due to non-specific binding of misfolded aggregates or host cell proteins (HCPs). Ensure thorough washing (increase wash cycles to 5-6) and optimize your blocking conditions. Use a blocking buffer containing 3-5% BSA or a commercial protein-free blocker. If using a His-tag for capture, imidazole (5-10 mM) in wash buffers can reduce non-specific binding. Always include a control from E. coli expressing an empty vector.
Q2: My Surface Plasmon Resonance (SPR) binding kinetics assay shows an abnormally high dissociation rate (Kd) for my refolded protein. What steps should I take? A: A high Kd often indicates improper folding or instability. First, verify correct disulfide bond formation using non-reducing SDS-PAGE. For cytoplasmic E. coli expression with enhanced disulfide formation systems (e.g., Origami or SHuffle strains), ensure the cytoplasm is sufficiently oxidizing; check that growth media contains the appropriate redox supplements. Purify the protein under non-reducing conditions and use a fresh, reducing agent-free running buffer for SPR. Analyze the sensorgram for bulk refractive index shifts, which may indicate aggregation.
Q3: The biological activity (specific activity) of my purified disulfide-bonded protein is low despite a high yield and good ELISA binding. Why? A: This discrepancy suggests the protein is immunologically detectable (correct epitopes) but not fully functionally folded. ELISA may recognize partially folded intermediates. Assess conformational homogeneity using analytical size-exclusion chromatography (SEC) or differential scanning fluorimetry (DSF). The functional assay may require specific post-translational modifications or cofactors not present in the E. coli cytoplasm. Re-optimize induction conditions (lower temperature, slower induction) to favor slow, correct folding.
Q4: How do I determine if my low binding affinity in kinetics assays is due to faulty protein or an assay artifact? A: Perform a ligand-binding activity test. If your protein is an enzyme, measure catalytic turnover (kcat). For non-enzymes, use an orthogonal method like isothermal titration calorimetry (ITC) to confirm binding thermodynamics. Ensure the immobilized ligand in your SPR/BLI assay is correctly oriented and not denatured. Always run a positive control with a commercially available, active protein if possible. Check for mass loss or degradation of your analyte via SDS-PAGE after the kinetics run.
Q5: When comparing proteins from different E. coli disulfide-engineered strains (e.g., SHuffle vs. Origami), what internal controls should I use for functional assays? A: Always include:
- A non-disulfide-bonded variant (Cys-to-Ser/Ala mutant) as a negative folding control.
- A commercially sourced active protein as an inter-assay positive control.
- A process control: Purify a native E. coli cytoplasmic protein (e.g., MBP) from each strain to control for strain-specific purification artifacts. Normalize all activity data to the concentration of properly folded protein as determined by a quantitative method like reverse-phase HPLC, not just total protein.
Table 1: Typical Performance Metrics for Functional Assays of Disulfide-Bonded Proteins from Engineered E. coli Strains
| Strain / System | Typical Soluble Yield (mg/L) | % Correct Disulfide Bond (by LC-MS) | Specific Activity (U/mg) | Apparent Kd (nM) by SPR | Common Assay Pitfall |
|---|---|---|---|---|---|
| BL21(DE3) (Standard Cytosol) | 5-20 | <10% | Low/Variable | >1000 | Aggregation, no activity |
| Origami (DE3) (trxB-/gor- mutant) | 10-50 | 40-70% | Moderate | 10-100 | Inconsistent oxidation |
| SHuffle T7 (DsbC in cytosol) | 15-80 | 60-90% | High | 1-20 | Protease sensitivity |
| CyDisCo System (Sulfhydryl Oxidase + PDI) | 30-100 | 70-95% | High | 1-10 | Cost, specialized media |
Table 2: Troubleshooting ELISA for Disulfide-Bonded Proteins: Signal Patterns & Solutions
| Observed Signal Pattern | Potential Root Cause | Recommended Action |
|---|---|---|
| High Background, Low Specific Signal | Non-specific binding of misfolded aggregates | Increase stringency of washes; change blocking buffer; pre-clear lysate |
| Low Signal Across All Samples | Denatured capture antibody; incorrect detection Ab | Check antibody pair compatibility; re-titer antibodies; confirm protein elution buffer is compatible |
| Signal in Negative Control | Incomplete blocking; cross-reactive antibodies | Use a different blocking agent (e.g., casein); include additional control (unrelated protein) |
| High Variability Between Replicates | Inconsistent plate washing or coating | Use automated washer; ensure consistent coating time/temp; check for plate defects |
Experimental Protocols
Protocol 1: Direct ELISA for Rapid Folding Assessment Purpose: To quickly compare the display of native-like epitopes in proteins purified from different E. coli strains or conditions.
- Coating: Dilute purified protein samples in PBS (pH 7.4) to 2-5 µg/mL. Add 100 µL/well to a 96-well high-binding plate. Incubate overnight at 4°C.
- Washing: Aspirate and wash plate 3x with 300 µL/well of PBST (PBS + 0.05% Tween-20).
- Blocking: Add 200 µL/well of blocking buffer (3% BSA in PBST). Incubate for 2 hours at room temperature (RT). Wash 3x with PBST.
- Primary Antibody: Add 100 µL/well of primary antibody (conformation-sensitive if available) diluted in blocking buffer. Incubate 1-2 hours at RT. Wash 5x with PBST.
- Secondary Antibody: Add 100 µL/well of HRP-conjugated secondary antibody diluted in blocking buffer. Incubate 1 hour at RT in the dark. Wash 5x with PBST.
- Detection: Add 100 µL/well of TMB substrate. Incubate for 5-15 minutes. Stop reaction with 50 µL/well of 2M H2SO4.
- Readout: Measure absorbance at 450 nm immediately.
Protocol 2: Determination of Binding Kinetics by Surface Plasmon Resonance (SPR) Purpose: To quantitatively measure the binding affinity (Kd) and kinetics (ka, kd) of a folded disulfide-bonded protein to its target.
- Ligand Immobilization: Dilute the target molecule (ligand) in 10 mM sodium acetate buffer (pH typically 4.0-5.5). Using a CMS chip, activate carboxyl groups with a 1:1 mixture of 0.4 M EDC and 0.1 M NHS for 7 minutes. Inject the ligand solution (typically 10-50 µg/mL) for 5-7 minutes to achieve ~50-100 RU of coupling. Deactivate excess esters with 1 M ethanolamine-HCl (pH 8.5) for 7 minutes.
- Analyte Preparation: Dialyze the purified disulfide-bonded protein (analyte) into SPR running buffer (e.g., PBS-P+, 0.01% P20 surfactant, pH 7.4). Centrifuge at 15,000 x g for 10 minutes before use to remove aggregates. Prepare a 2-fold dilution series (e.g., 0.5x, 1x, 2x, 4x, 8x estimated Kd).
- Kinetics Experiment: Prime the system with running buffer. Inject analyte samples over the ligand and reference surfaces for 3-5 minutes (association phase), followed by running buffer for 5-10 minutes (dissociation phase) at a flow rate of 30 µL/min. Regenerate the surface with a 30-second pulse of glycine-HCl (pH 2.0-2.5) if necessary.
- Data Analysis: Subtract the reference cell signal and buffer blank injections. Fit the double-referenced sensograms to a 1:1 binding model using the SPR instrument’s software (e.g., Biacore Evaluation Software). Report ka (association rate, M⁻¹s⁻¹), kd (dissociation rate, s⁻¹), and KD (kd/ka, M).
Visualizations
Title: Workflow for Correlating Folding & Activity
Title: SPR Binding Kinetics Assay Cycle
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Functional Assays of Disulfide-Bonded Proteins
| Reagent / Material | Function & Rationale | Example Product / Note |
|---|---|---|
| Engineered E. coli Strains (SHuffle, Origami, CyDisCo) | Provide an oxidizing cytoplasm or disulfide bond catalysts to promote correct folding in vivo. | NEB SHuffle T7, Novagen Origami B. |
| Non-Reducing Lysis & Wash Buffers | Maintain existing disulfide bonds during cell disruption and purification. | Include 5-10 mM EDTA, avoid DTT/BME. |
| Conformation-Sensitive Antibodies | Detect specific, natively folded epitopes in ELISA, distinguishing from misfolded protein. | Often monoclonal; require careful validation. |
| High-Binding ELISA Plates | Maximize adsorption of often hydrophobic, disulfide-bonded proteins for immunoassay. | Corning Costar 9018 or equivalent. |
| SPR Sensor Chips (CMS Series) | Gold surface with carboxymethyl dextran for covalent ligand immobilization via amine coupling. | Cytiva Series S Chip CMS. |
| P20 Surfactant | Reduces non-specific binding in SPR running buffers, critical for hydrophobic proteins. | Cytiva BR-1000-54. |
| HRP-Conjugated Antibodies & TMB Substrate | Standard, sensitive detection system for colorimetric ELISA. | Commercial pairs recommended. |
| Size-Exclusion Chromatography (SEC) Column | Assess protein aggregation state and monodispersity post-purification. | Superdex 75 or 200 Increase columns. |
| Redox Buffers (GSH/GSSG) | Used in refolding or in-vitro oxidation assays to test disulfide formation efficiency. | Prepare fresh; specific ratios critical. |
Technical Support Center
This support center addresses common technical issues encountered when using the SHuffle, Origami, and CyDisCo E. coli strains for cytoplasmic disulfide bond formation.
Troubleshooting Guides & FAQs
Q1: My target protein expresses in SHuffle but is entirely insoluble. What should I check? A: This is a common issue. Follow this checklist:
- Temperature: Reduce expression temperature to 16-25°C immediately after induction. SHuffle's oxidizing pathway is more active at lower temperatures.
- Induction Timing: Induce at a lower OD600 (0.4-0.6) to reduce metabolic burden.
- Lysis Buffer: Ensure your lysis buffer contains 10-20 mM N-Ethylmaleimide (NEM) to alkylate free thiols and "trap" the oxidation state during cell disruption.
- Plasmid Verification: Confirm your plasmid uses a T7 promoter compatible with SHuffle's DE3 lysogen.
Q2: I see no yield improvement with CyDisCo despite co-expressing DsbC. What are the potential causes? A: CyDisCo requires precise stoichiometry.
- Promoter Strength: Verify that the sulfhydryl oxidase (e.g., Erv1p) and disulfide isomerase (DsbC) are expressed from weaker promoters than your target protein. Use a plasmid with inducible, titratable promoters (e.g., pBAD) for the helper proteins.
- Strain Background: The parent strain (e.g., BL21) must be trxB-/gor- for CyDisCo to be effective. Confirm the genotype.
- Control Experiment: Perform a control expression with the empty helper plasmid to isolate the impact of the CyDisCo system.
Q3: My Origami strain shows very slow growth post-transformation. Is this normal? A: Yes, but it can be managed. Origami (Δgor ΔtrxB) has a severe reducing pathway deletion, leading to slow growth and sensitivity to oxygen.
- Media Supplementation: Always supplement media with 0.5-1 mM IPTG for selection (due to the lacY mutation on the chromosome) and add 5 mM sodium succinate as an additional carbon source to boost growth.
- Antibiotics: Use kanamycin (for the trxB mutation) and tetracycline (for the gor mutation) for selection and maintenance, not just ampicillin for the plasmid.
- Growth Time: Allow 36-48 hours for colony formation on plates at 37°C.
Q4: How do I troubleshoot low fidelity (mispaired disulfides) in my final product from any of these systems? A: Low fidelity indicates a bottleneck in isomerization.
- Isomerase Cofactor: For SHuffle, ensure the media contains heme precursors (add 50 µM FeCl3 or use rich media) for functional DsbC. For CyDisCo, the same applies if using Erv1p.
- Post-Lysis Redox: Add a redox buffer (e.g., 10 mM reduced/1 mM oxidized glutathione) to your purification buffers to allow disulfide shuffling in vitro.
- Analytical Method: Use a non-reducing vs. reducing SDS-PAGE assay coupled with mass spectrometry to identify scrambled species.
Quantitative Comparison Table
| Metric | SHuffle T7 | Origami B | CyDisCo (in BL21) | Notes |
|---|---|---|---|---|
| Genetic Modifications | Δgor ΔtrxB, dsbC expressed in cytosol, DE3 lysogen | Δgor ΔtrxB, lacY, DE3 lysogen | Parent strain (e.g., BL21) + plasmid(s) expressing sulfhydryl oxidase (Erv1p) & DsbC | SHuffle provides isomerase natively. CyDisCo is plasmid-based. |
| Typical Yield (mg/L) | 5-50 | 1-20 | 10-100+ | Highly target-dependent. CyDisCo often reports the highest yields. |
| Growth Speed | Moderate | Slow | Fast (depends on parent strain) | Origami is slow due to severe redox mutations. |
| Oxidation Fidelity | High | Low-Moderate | High | SHuffle & CyDisCo provide isomerase activity (DsbC). Origami only provides an oxidizing environment. |
| Key Requirement | Heme for DsbC, Low temp induction | Succinate in media, Dual antibiotic selection | Balanced co-expression of helper proteins | |
| Best For | Proteins requiring isomerization; complex disulfides. | Simple 1-2 disulfide bonds; when plasmid-based systems are undesired. | High-yield production of complex, multi-disulfide proteins. |
Key Experimental Protocols
Protocol 1: Expression Test for Disulfide Bond Formation
- Transform your target plasmid into SHuffle, Origami, and a CyDisCo-equipped BL21 strain. Plate on appropriate antibiotic plates.
- Inoculate 5 mL starter cultures and grow overnight at 30°C (SHuffle, CyDisCo) or 37°C (Origami).
- Dilute 1:100 into fresh media with antibiotics (and supplements: 5mM succinate for Origami, 50 µM FeCl3 for SHuffle). Grow at 37°C to OD600 ~0.6.
- Induce with optimal inducer (e.g., 0.1-1 mM IPTG). Critical Step: Shift SHuffle cultures to 25°C or 16°C. Origami and CyDisCo can be expressed at 30°C.
- Harvest cells 4-16 hours post-induction by centrifugation.
- Analyze by Non-Reducing vs. Reducing SDS-PAGE. Resuspend pellet in lysis buffer with and without β-mercaptoethanol/DTT. A mobility shift indicates disulfide formation.
Protocol 2: Assessing Solubility and Fidelity
- Follow Protocol 1 for expression.
- Lysis: Lyse cells (sonication/lysozyme) in Buffer A (50 mM Tris, 150 mM NaCl, pH 8.0, 20 mM NEM, 1 mM PMSF).
- Centrifuge at 15,000 x g for 30 min to separate soluble (supernatant) and insoluble (pellet) fractions.
- Analyze Fractions: Run soluble and resuspended pellet samples on reducing SDS-PAGE to determine solubility ratio.
- Fidelity Check: Purify the soluble protein via IMAC. Perform LC-MS on the intact protein under non-reducing conditions to compare observed mass with theoretical mass for correct disulfide pairing.
Signaling Pathway & Workflow Diagrams
Title: SHuffle T7 Strain Protein Folding Workflow
Title: Cytoplasmic Disulfide Bond Formation Pathways
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function & Rationale |
|---|---|
| N-Ethylmaleimide (NEM) | Thiol-alkylating agent. Added to lysis buffer to "freeze" the redox state of cysteines during cell disruption, preventing artificial oxidation/reduction. |
| Sodium Succinate | Carbon source supplement. Crucial for improving the poor growth of gor-/trxB- strains like Origami by entering the TCA cycle. |
| Ferric Chloride (FeCl₃) | Heme precursor. Required for functional holo-DsbC in the SHuffle cytoplasm and for the Erv1p sulfhydryl oxidase in CyDisCo. |
| Redox Buffers (GSH/GSSG) | Glutathione redox couples. Used in in vitro refolding or purification buffers to maintain a specific redox potential and allow disulfide reshuffling. |
| Dual Antibiotic Plates (Kan/Tet) | Selection plates for Origami strains. Necessary to maintain the genomic trxB (KanR) and gor (TetR) mutations. |
| Non-Reducing SDS-PAGE Sample Buffer | Diagnostic tool. Lacks DTT/β-mercaptoethanol, allowing visualization of disulfide-bonded protein complexes and mobility shifts. |
COMPARATIVE ANALYSIS OF METABOLIC BURDEN AND GROWTH CHARACTERISTICS ACROSS ENGINEERED STRAINS
Technical Support Center
This support center provides troubleshooting and FAQs for researchers measuring metabolic burden and growth in E. coli strains engineered for enhanced cytoplasmic disulfide bond formation. All content is framed within the thesis context: "Enhancing disulfide bond formation in E. coli cytoplasm."
Troubleshooting Guides
Issue 1: Engineered strain exhibits severely impaired growth rate post-induction.
- Symptoms: After inducing the disulfide bond-forming machinery (e.g., DsbC expression) and/or target protein, the culture optical density (OD600) stops increasing or increases very slowly.
- Potential Causes & Solutions:
- Cause: Extreme metabolic burden from concurrent high-level expression of multiple system components (e.g., sulfhydryl oxidase, disulfide isomerase, target protein, and reductase knockout).
- Solution: Titrate inducer concentration. Use a lower IPTG or arabinose concentration to reduce expression load. Refer to Table 1 for comparative data.
- Cause: Redox imbalance leading to toxic accumulation of reactive oxygen species or depletion of reducing equivalents (NADPH).
- Solution: Supplement media with 0.5% w/v glucose or 1mM succinate as an additional energy source. Monitor dissolved oxygen to avoid hypoxia.
- Cause: Incompatibility between the chosen promoter strength and the genetic background (e.g., ΔtrxB gor).
- Solution: Switch to a weaker or tunable promoter (e.g., PBAD, Ptet) for expression system components.
- Cause: Extreme metabolic burden from concurrent high-level expression of multiple system components (e.g., sulfhydryl oxidase, disulfide isomerase, target protein, and reductase knockout).
Issue 2: High target protein yield but low fraction of correctly folded, soluble product.
- Symptoms: SDS-PAGE shows a strong band at the expected molecular weight, but solubility assay (comparison of total vs. soluble lysate) shows majority in inclusion bodies. Activity assay is low.
- Potential Causes & Solutions:
- Cause: Disulfide bond formation machinery is overwhelmed or improperly balanced. Oxidase activity may outpace isomerase activity.
- Solution: Co-express a sulfhydryl oxidase (e.g., Erv1p) and a disulfide isomerase (e.g., DsbC) on separate plasmids with independently tunable promoters to optimize their ratio.
- Cause: Protein folding is too slow relative to aggregation, despite correct disulfide formation.
- Solution: Lower the cultivation temperature to 25-30°C post-induction to slow synthesis and favor folding. Consider co-expression of a relevant chaperone (e.g., DnaK-DnaJ-GrpE or GroEL-GroES).
- Cause: Disulfide bond formation machinery is overwhelmed or improperly balanced. Oxidase activity may outpace isomerase activity.
Issue 3: Inconsistent results between replicates when measuring growth parameters.
- Symptoms: High variability in doubling time, maximum OD600, or lag phase duration between biological replicates in microplate or flask cultures.
- Potential Causes & Solutions:
- Cause: Inoculum history effect. Pre-culture growth phase impacts metabolic state.
- Solution: Always inoculate main culture from pre-cultures harvested at the same mid-exponential phase (OD600 ~0.6-0.8). Use a consistent pre-culture volume for inoculation (e.g., 1:100 dilution).
- Cause: Inadequate aeration in small-scale cultures, especially with metabolically burdened strains.
- Solution: For deep-well plates, use a shaking speed ≥900 rpm. For baffled flasks, ensure culture volume is ≤10-20% of total flask volume.
- Cause: Inoculum history effect. Pre-culture growth phase impacts metabolic state.
Frequently Asked Questions (FAQs)
Q1: What are the most informative metrics to quantify metabolic burden for my engineered disulfide bond strains? A: The core metrics are: (1) Specific Growth Rate (μ) during exponential phase, (2) Maximum Biomass Yield (OD600 max), (3) Lag Phase Duration, and (4) Product Yield per Cell (e.g., mg protein per OD600 unit). Comparing these metrics between your engineered strain, a control strain (empty vector), and the parental wild-type strain under identical conditions is essential. See Table 1.
Q2: Should I use minimal or rich media for these comparative growth studies? A: Rich media (e.g., LB, TB) is better for initial characterization as it supports fast growth, making burden effects more pronounced and experiments quicker. Minimal media (e.g., M9) is crucial for assessing the strain's metabolic efficiency and for industrial relevance, as burden effects are often more severe due to the need for precursor synthesis.
Q3: My engineered strain with a trxB/gor knockout grows very poorly. How can I improve robustness? A: Consider using suppressor mutations (e.g., ahpC) that partially restore the redox balance, or use attenuated knockout strains (e.g., ΔtrxB* alone or gorts). Always compare the burden of the full knockout versus attenuated backgrounds. Expression of a cytoplasmically adapted disulfide isomerase (e.g., DsbCcyt) is often still required in these strains.
Q4: How do I decouple the metabolic burden of the disulfide system from the burden of my target protein expression? A: Perform a three-way comparison: (A) Host strain with empty vector, (B) Host strain expressing only the disulfide system components (no target gene), (C) Host strain expressing both the system and the target protein. The burden from the system alone is (B-A). The additional burden from the target is (C-B).
Data Presentation
Table 1: Comparative Growth Metrics of E. coli Strains Engineered for Cytoplasmic Disulfide Bond Formation Conditions: LB medium, 37°C, induced with 0.1 mM IPTG at OD600=0.3. Data is representative.
| Strain & Description | Specific Growth Rate, μ (h⁻¹) | Max OD600 | Lag Phase (min) | Soluble Target Yield (mg/L/OD) |
|---|---|---|---|---|
| BW25113 (Wild-type control) | 0.95 ± 0.05 | 4.8 ± 0.2 | 30 ± 5 | N/A |
| BW25113 + pEmpty Vector | 0.92 ± 0.04 | 4.6 ± 0.3 | 35 ± 8 | N/A |
| BW25113 ΔtrxB gor (SHuffle T7 genotype) | 0.45 ± 0.06 | 2.1 ± 0.3 | 120 ± 15 | N/A |
| ΔtrxB gor + pDsbCcyt (system only) | 0.40 ± 0.05 | 1.9 ± 0.2 | 135 ± 20 | N/A |
| ΔtrxB gor + pDsbCcyt + pTarget-Protein | 0.25 ± 0.04 | 1.2 ± 0.2 | 180 ± 25 | 15 ± 3 |
| ΔtrxB gor ahpC* + pDsbCcyt + pTarget-Protein (Suppressor) | 0.35 ± 0.05 | 1.8 ± 0.3 | 110 ± 20 | 22 ± 4 |
Experimental Protocols
Protocol 1: High-Throughput Growth Curve Analysis in a Microplate Reader
- Inoculum Prep: Grow overnight cultures of all strains to be compared in 2 mL of appropriate medium with antibiotics.
- Dilution: Dilute overnight culture 1:100 into fresh, pre-warmed medium (with antibiotics, ± inducer as required for baseline).
- Plate Setup: Transfer 200 µL of each diluted culture into at least 4 replicate wells of a sterile, clear flat-bottom 96-well plate. Include wells with medium only as a blank.
- Measurement: Place plate in a pre-warmed (37°C) microplate reader with continuous linear shaking. Measure OD600 every 10 minutes for 12-24 hours.
- Analysis: Subtract blank OD values. Calculate the specific growth rate (μ) from the linear region of the ln(OD) vs. time plot.
Protocol 2: Measuring Soluble vs. Insoluble Protein Fraction
- Harvest: Induce expression as required. After suitable time, harvest cells from 1 mL culture by centrifugation (13,000 x g, 2 min, 4°C). Record OD600 of culture.
- Lysis: Resuspend pellet in 100 µL BugBuster Master Mix or Lysozyme/Triton lysis buffer. Incubate with gentle mixing for 20 min at room temperature.
- Separation: Centrifuge lysate at 15,000 x g for 20 min at 4°C. Carefully transfer the supernatant (soluble fraction) to a new tube.
- Wash & Solubilize: Resuspend the pellet (insoluble fraction) in 100 µL of 1x SDS-PAGE loading buffer. Denature the supernatant sample by adding 25 µL of 5x SDS-PAGE loading buffer.
- Analysis: Boil all samples for 10 min, then load equal volumes (e.g., 10-20 µL) or volumes normalized by original OD600 onto an SDS-PAGE gel for Coomassie staining or Western blot.
Mandatory Visualization
Diagram Title: Metabolic Burden Sources and Outcomes in Engineered E. coli
Diagram Title: Experimental Workflow for Comparative Growth Analysis
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Disulfide Bond/Burden Research |
|---|---|
| SHuffle T7 Express E. coli | Commercial strain (ΔtrxB gor ahpC* + cytoplasmic DsbC) providing a robust starting platform for cytoplasmic disulfide bond formation. |
| pBAD-based Expression Vectors | Allows fine-tuning of expression level for disulfide system components using arabinose, helping to minimize burden. |
| BugBuster HT Protein Extraction Reagent | Efficient, scalable chemical lysis reagent for parallel processing of many samples to separate soluble/insoluble protein fractions. |
| Resazurin Sodium Salt | Redox-sensitive dye used in alamarBlue assays to measure cellular metabolic activity and viability as a proxy for burden. |
| NADP/NADPH Assay Kit (Colorimetric) | Quantifies the ratio of NADP+ to NADPH, directly assessing the redox cofactor imbalance caused by disulfide engineering. |
| Pierce Ellman's Reagent (DTNB) | Measures free sulfhydryl groups, useful for assessing the oxidative state of cysteine residues in expressed target proteins. |
| Tunair Flask or 96-Well Microplate | Provides optimal, consistent aeration for growth studies of oxygen-sensitive, metabolically burdened strains. |
Technical Support Center: Troubleshooting Guides and FAQs
This support center is designed to assist researchers working within the thesis context of Enhancing disulfide bond formation in E. coli cytoplasm. It addresses common experimental hurdles in selecting and optimizing systems for expressing disulfide-bonded proteins.
FAQs and Troubleshooting
Q1: My protein of interest contains multiple disulfide bonds and is not expressing solubly in the standard SHuffle T7 strain. What should I try next?
A: The SHuffle strain is engineered for cytoplasmic disulfide bond formation by constitutively expressing a truncated dsbC gene and lacking trxB and gor to create an oxidizing cytoplasm. For proteins with high disulfide complexity, consider these steps:
- Lower Growth Temperature: Reduce expression temperature to 20-25°C post-induction to slow protein synthesis and favor correct folding.
- Tune Expression: Use a lower concentration of inducer (e.g., 0.1 mM IPTG) to reduce the rate of protein production.
- Strain Upgrade: Move to a more specialized strain like SHuffle B or C, which also carry mutations to enhance disulfide bond isomerization (dsbC is co-expressed with its specific reductase, dsbD).
- Protocol Check: Ensure you are using the recommended media (e.g., rich media like 2xYT) and that antibiotics are fresh.
Q2: I am comparing Origami B and SHuffle strains. When should I choose one over the other for cytoplasmic expression?
A: The choice hinges on the need for isomerase activity. See the comparison table below.
| Feature | Origami B DE3 | SHuffle T7 Express |
|---|---|---|
| Genotype | Δgor ΔtrxB, cytoplasmic | Δgor ΔtrxB, cytoplasmic |
| Key Enzyme | Thioredoxin reductase & glutathione reductase knocked out. | Truncated dsbC (disulfide isomerase) targeted to the cytoplasm. |
| Primary Strength | Strongly oxidizing cytoplasm for bond formation. | Oxidizing cytoplasm plus isomerase activity to shuffle incorrect bonds. |
| Best For | Proteins with simple, known disulfide connectivity. | Proteins with complex or unknown disulfide bond patterns requiring rearrangement. |
| Common Issue | May form aggregates if bonds form incorrectly without isomerization. | Slower growth than wild-type; requires careful monitoring of culture density. |
Q3: My downstream need is high-throughput screening of antibody fragments. Which system offers the best balance of speed and correct folding?
A: For screening, BL21(DE3) pLysS transformed with a plasmid co-expressing DsbC is often optimal. This system provides:
- Rapid Growth: BL21 background grows faster than knockout strains.
- Controlled Expression: pLysS provides tighter repression for toxic proteins.
- On-demand Isomerase: DsbC is expressed from the same plasmid as your target, ensuring it's available when needed.
- Protocol: Co-transform with both plasmids, select with both antibiotics, and induce with a low dose of IPTG (0.05-0.2 mM) at mid-log phase at 25°C.
Q4: I have confirmed soluble expression, but my purified protein is inactive. How can I troubleshoot disulfide bond correctness?
A: This suggests possible misfolding or non-native disulfide bonds.
- Run a Non-Reducucing vs. Reducing SDS-PAGE: Compare migration. A faster migration under non-reducing conditions typically indicates intra-chain disulfide formation. No shift may suggest no bonds or inter-chain bonds.
- Mass Spec Analysis: Perform peptide mapping under non-reducing conditions to identify which cysteines are linked.
- Optimize Redox Buffer: In your lysis and purification buffers, include a low-molecular-weight redox couple like GSH:GSSG (e.g., 5:1 ratio). This can help "refold" or correct mispaired bonds in vitro.
- Switch Strains: If using Origami, move to a SHuffle strain for its in vivo isomerase activity.
Key Experimental Protocols
Protocol 1: Small-Scale Expression Test in Multiple Strains
- Transform your target plasmid into SHuffle T7, Origami B, and a control strain (e.g., BL21).
- Inoculate 5 mL of LB (+ appropriate antibiotics) with a single colony. Grow overnight at 30°C (for SHuffle/Origami) or 37°C (BL21).
- Dilute 1:100 into 5 mL fresh medium in a 50 mL tube. Grow to OD600 ~0.6.
- Induce with 0.5 mM IPTG (or optimal concentration for your vector).
- Express for 4-6 hours at 30°C or 16-20 hours at 18°C.
- Harvest cells by centrifugation. Resuspend in 500 µL lysis buffer (e.g., PBS with 1 mg/mL lysozyme, optional protease inhibitors).
- Lyse by sonication or freeze-thaw.
- Separate soluble and insoluble fractions by centrifugation (14,000 rpm, 20 min).
- Analyze total, soluble, and pellet fractions by SDS-PAGE (run both reducing and non-reducing gels).
Protocol 2: Assessing Disulfide Bond Formation via SDS-PAGE
- Prepare protein samples in Non-Reducing Sample Buffer (Laemmli buffer without β-mercaptoethanol or DTT).
- Prepare duplicate samples in Reducing Sample Buffer (Laemmli buffer with 5% β-mercaptoethanol).
- Do not boil non-reduced samples, or boil for <2 min, to avoid artificial disulfide scrambling. Reduced samples can be boiled for 5 min.
- Run all samples on the same polyacrylamide gel.
- Compare migration. Correct intramolecular disulfides cause a compact structure and faster migration in the non-reducing lane.
Visualization: System Selection Logic
Decision Workflow for Disulfide Bond Expression System
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Research |
|---|---|
| SHuffle T7 Express E. coli | Specialized strain for cytoplasmic expression with disulfide bond isomerase (DsbC) activity. |
| Origami B(DE3) E. coli | Strain with thioredoxin and glutathione reductase knockouts, creating an oxidizing cytoplasm. |
| pET-39b(+) Vector | Expression vector containing the DsbA signal sequence for periplasmic targeting; can be modified for cytoplasmic studies. |
| pACYCDuet-1 Vector | A low-copy number plasmid ideal for co-expressing chaperones or foldases (like DsbC) alongside the target protein. |
| Reduced (GSH) & Oxidized (GSSG) Glutathione | Used in redox buffers to fine-tune the oxidation potential of lysis or refolding buffers in vitro. |
| β-mercaptoethanol / DTT | Reducing agents for preparing control samples in SDS-PAGE to break disulfide bonds. |
| Iodoacetamide | Alkylating agent used to block free cysteines and prevent disulfide scrambling during sample prep for MS. |
| Anti-DsbC Antibody | Useful for monitoring the expression of the DsbC isomerase in engineered strains via Western blot. |
| 2xYT Media | Nutrient-rich growth medium often recommended for robust growth of slower-growing oxidative strains. |
Conclusion
Enhancing disulfide bond formation in the E. coli cytoplasm is no longer an insurmountable barrier but a tractable engineering challenge. By understanding the foundational redox biology, implementing robust methodological toolkits, systematically troubleshooting production issues, and rigorously validating outcomes, researchers can reliably produce complex disulfide-bonded proteins. The comparative success of platforms like SHuffle and CyDisCo demonstrates the field's maturity, offering viable alternatives to periplasmic or mammalian systems for many therapeutics. Future directions point toward more dynamic redox control systems, integration with AI-driven folding prediction, and extension to novel protein formats. This capability significantly expands E. coli's role as a versatile and cost-effective host for next-generation biologics, accelerating preclinical research and development pipelines in biomedicine.