Unfolded Protein Response (UPR): From ER Stress Mechanisms to Therapeutic Targeting in Disease
This article provides a comprehensive overview of the Endoplasmic Reticulum (ER) unfolded protein response (UPR), a critical signaling network that maintains cellular proteostasis.
Unfolded Protein Response (UPR): From ER Stress Mechanisms to Therapeutic Targeting in Disease
Abstract
This article provides a comprehensive overview of the Endoplasmic Reticulum (ER) unfolded protein response (UPR), a critical signaling network that maintains cellular proteostasis. We detail the molecular mechanisms of the three UPR sensors—PERK, IRE1α, and ATF6—and their dual roles in adaptive survival and pro-apoptotic signaling. The content explores the established and emerging methodologies for detecting UPR activation, addresses common challenges in experimental analysis, and validates UPR's pathophysiological significance in cancer, neurodegenerative disorders, metabolic diseases, and immune dysregulation. Aimed at researchers and drug development professionals, this review synthesizes current knowledge on UPR as a promising therapeutic target, highlighting preclinical inhibitors and context-dependent modulation strategies for novel treatments.
The Core Machinery of the UPR: Understanding ER Stress Sensors and Signaling Pathways
The Endoplasmic Reticulum: Guardian of Cellular Proteostasis
The endoplasmic reticulum (ER) is the largest membrane-bound organelle in eukaryotic cells, functioning as a primary site for the synthesis, folding, and modification of secretory and transmembrane proteins. Approximately one-third of all cellular proteins are processed within this compartment [1]. The ER also serves as the major intracellular calcium store, contributes to lipid and steroid synthesis, and participates in carbohydrate metabolism [2] [1]. This multifunctional organelle maintains a specialized environment within its lumen, characterized by a high concentration of chaperone proteins (such as BiP/GRP78), calcium-dependent foldases, and oxidoreductases that collectively ensure proper protein folding and quality control [3] [1].
ER homeostasis refers to the finely balanced state where the protein-folding capacity of the ER meets the cellular demand for protein synthesis and processing. Maintaining this equilibrium is crucial for cellular function, as its disruption has been implicated in the pathogenesis of numerous human diseases, including neurodegenerative disorders, diabetes, cancer, and inflammatory conditions [3] [2] [4].
Triggers of ER Stress: Disruption of Proteostasis
ER stress occurs when the accumulation of unfolded or misfolded proteins in the ER lumen exceeds the organelle's processing capacity, disrupting proteostasis [2] [5]. This imbalance activates a sophisticated signaling network known as the unfolded protein response (UPR) [3] [6].
The triggers of ER stress are diverse and can be categorized as follows:
Table 1: Major Inducers of Endoplasmic Reticulum Stress
| Stress Category | Specific Inducers | Mechanism of Action | Pathophysiological Context |
|---|---|---|---|
| Disturbances in ER Calcium Homeostasis | Thapsigargin (SERCA pump inhibitor) | Depletes ER calcium stores, impairing calcium-dependent chaperones [2] [7] | Experimental models & potential disease mechanisms |
| Impaired Protein Glycosylation | Tunicamycin (N-linked glycosylation inhibitor) | Causes accumulation of unglycosylated, misfolded proteins [2] [8] | Experimental models & congenital disorders of glycosylation |
| Oxidative Stress | Reactive Oxygen Species (ROS) | Disrupts disulfide bond formation, damaging proteins and ER membrane [3] [5] | Metabolic diseases, neurodegeneration, aging |
| Nutrient Deprivation | Glucose deprivation, energy/ATP depletion | Reduces energy for chaperone function and protein folding [2] [5] | Ischemia, solid tumor microenvironment |
| Genetic Mutations | Mutations in secretory proteins (e.g., mutant insulin) | Production of intrinsically misfolded proteins that overwhelm quality control [2] [5] | Genetic diseases like Wolcott-Rallison syndrome, diabetes |
| Lipid Homeostasis Perturbation | Saturated fatty acids (e.g., palmitic acid) | Alters ER membrane composition, sensed by UPR transmembrane domains [2] [6] | Obesity, metabolic syndrome, atherosclerosis |
| Increased Protein Synthesis Demand | Viral infection, enhanced secretory pathway activity | Overwhelms ER folding capacity, creating a "folding bottleneck" [2] [5] | Viral pathogenesis, plasma cell differentiation |
The UPR is activated not only by protein-centric stresses but also through direct sensing of perturbations in lipid homeostasis via the transmembrane domains of UPR sensors, independently of their luminal sensing domains [6]. This demonstrates the ER's role as a comprehensive integrator of diverse cellular stress signals.
The Unfolded Protein Response: Molecular Mechanisms and Signaling Pathways
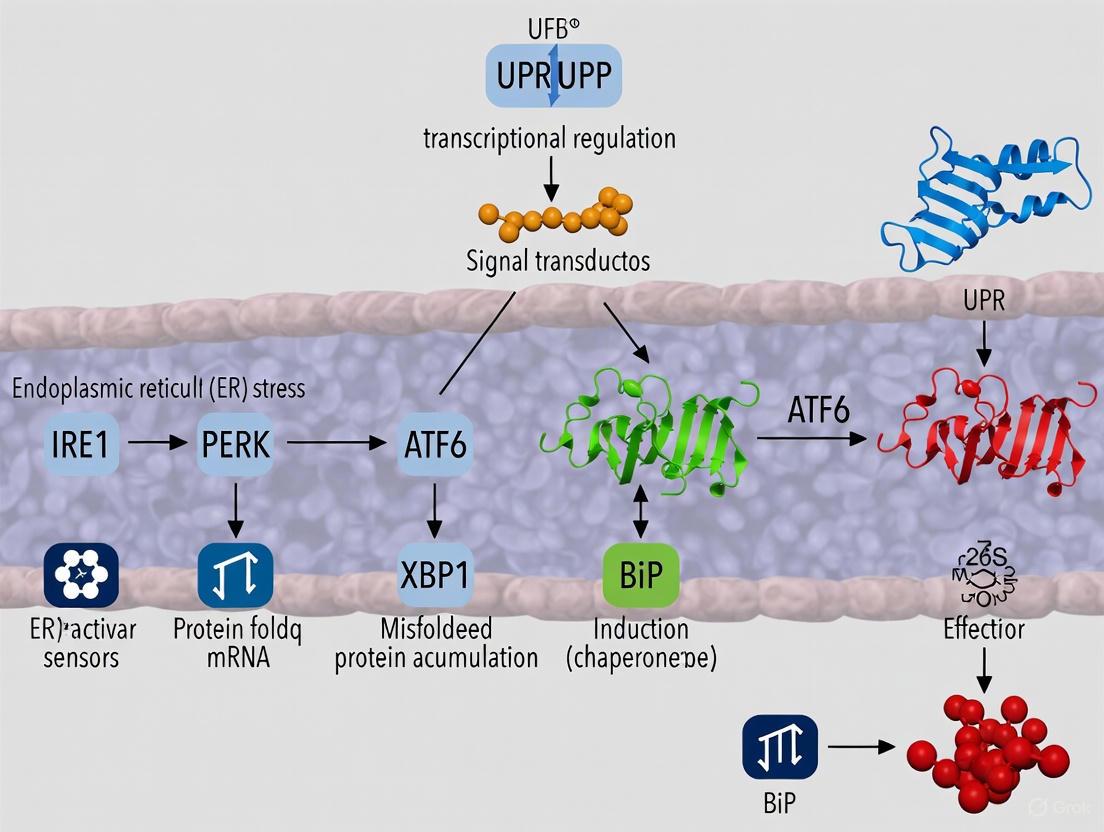
The UPR is orchestrated by three ER-transmembrane sensor proteins: PERK, IRE1α, and ATF6. Under non-stress conditions, these sensors are maintained in an inactive state through association with the ER chaperone BiP [3] [4]. The accumulation of unfolded proteins triggers BiP dissociation, leading to the activation of these sensors and the initiation of adaptive signaling cascades [3].
UPR Signaling Pathways: The three arms of the unfolded protein response and their downstream outcomes.
The PERK-eIF2α Pathway
PERK is a serine/threonine kinase that, upon activation, undergoes homodimerization and autophosphorylation. Its primary substrate is the α-subunit of eukaryotic translation initiation factor 2 (eIF2α). Phosphorylation of eIF2α (p-eIF2α) globally attenuates protein translation, reducing the incoming protein load on the stressed ER [3] [4]. However, this phosphorylation simultaneously enables the selective translation of specific mRNAs, notably that of activating transcription factor 4 (ATF4). ATF4 upregulates genes involved in amino acid metabolism, antioxidant responses, and ER chaperone expression [3] [2]. Under prolonged ER stress, ATF4 induces the expression of the pro-apoptotic transcription factor CHOP, which promotes cell death by downregulating anti-apoptotic proteins and increasing oxidative stress [3] [4].
The IRE1α-XBP1 Pathway
IRE1α is the most evolutionarily conserved UPR sensor, possessing both kinase and endoribonuclease activities. Its activation leads to the unconventional splicing of a 26-nucleotide intron from X-box binding protein 1 (XBP1) mRNA [3] [2]. This splicing event results in a frameshift, producing a stable and potent transcription factor, spliced XBP1 (XBP1s). XBP1s translocates to the nucleus and drives the expression of genes encoding ER chaperones, ER-associated degradation (ERAD) components, and lipid biosynthetic enzymes, thereby expanding the ER's protein-folding and processing capacity [3] [5]. Under irremediable ER stress, IRE1α can also initiate regulated IRE1-dependent decay (RIDD), degrading ER-localized mRNAs to further reduce protein load, and can promote apoptosis through JNK signaling [3] [4].
The ATF6 Pathway
ATF6 is a type II transmembrane protein that acts as a transcription factor. Upon ER stress, it translocates to the Golgi apparatus where it undergoes proteolytic cleavage by site-1 and site-2 proteases (MBTPS1/2) [3] [4]. This cleavage releases its cytosolic domain (ATF6f), which functions as an active transcription factor. ATF6f translocates to the nucleus and enhances the expression of ER chaperones (including BiP itself) and components of the ERAD machinery, acting in concert with XBP1s to restore ER homeostasis [3] [2].
Experimental Protocols for ER Stress Research
In Vitro Induction and Assessment of ER Stress
The following protocol details a standard approach for inducing and analyzing ER stress in mammalian cell lines, incorporating key assays for monitoring UPR activation.
Table 2: Key Research Reagents for ER Stress Studies
| Reagent Name | Category | Mechanism of Action | Common Working Concentration |
|---|---|---|---|
| Thapsigargin (Tg) | ER Ca²⁺ disruptor | Inhibits SERCA pumps, depleting ER calcium stores [2] [9] | 100-500 nM [2] [9] |
| Tunicamycin (Tm) | Protein glycosylation inhibitor | Blocks N-linked glycosylation, causing misfolded protein accumulation [2] [8] | Varies by cell type (e.g., 1-10 µg/mL) [8] |
| Brefeldin A | Protein transport inhibitor | Disrupts Golgi apparatus, blocking protein secretion from ER [1] | 1-10 µM |
| Dithiothreitol (DTT) | Reducing agent | Reduces disulfide bonds, preventing proper protein folding [1] | 1-5 mM |
| STF-083010 | IRE1α RNase inhibitor | Specifically inhibits IRE1α's endoribonuclease activity [2] | 10-100 µM [2] |
| ISRIB | PERK pathway inhibitor | Reverses eIF2α phosphorylation effects, restores translation [4] | 100-500 nM |
| 4-PBA / TUDCA | Chemical chaperones | Improves overall protein folding capacity, alleviates ER stress [1] [5] | 0.1-1 mM (TUDCA) [1] |
Protocol: Thapsigargin-Induced ER Stress in Endocrine Cell Lines [9]
Cell Culture and Seeding: Maintain relevant cell lines (e.g., MIN6 β-cells, αTC1-6 α-cells) in their appropriate growth media. Seed cells into 6-well culture plates and allow them to reach 70-80% confluency.
ER Stress Induction:
- Prepare a stock solution of thapsigargin in DMSO and dilute it in pre-warmed culture medium to the final desired concentration (e.g., 100 nM for MIN6 cells, 500 nM for αTC1-6 cells) [9].
- Replace the cell culture medium with the thapsigargin-containing medium. Include control wells treated with an equivalent volume of DMSO vehicle (e.g., 0.1% DMSO).
- Incubate cells for a predetermined time course (e.g., 6 hours and 24 hours) to capture both early and sustained UPR responses.
RNA Isolation and Quantitative PCR (RT-qPCR) Analysis:
- Lyse cells directly in the culture plate using a commercial lysis buffer containing β-mercaptoethanol.
- Isolate total RNA using a phenol-chloroform-based method (e.g., TRIzol) or a silica-membrane column system.
- Synthesize cDNA from 500 ng of total RNA using reverse transcriptase and random hexamer primers.
- Perform qPCR using primers for canonical UPR target genes:
- BiP/GRP78: A master regulator chaperone induced by all UPR arms.
- CHOP: A key pro-apoptotic transcript primarily regulated by the PERK-ATF4 axis.
- XBP1s: The spliced, active form specifically generated by IRE1α activation.
- Normalize expression data to housekeeping genes (e.g., Actin, GAPDH) and analyze using the ΔΔCt method to determine fold-changes relative to DMSO-treated controls.
Protein Extraction and Western Blot Analysis:
- Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors.
- Quantify protein concentration using a standard assay (e.g., Bradford).
- Separate 20-30 µg of total protein by SDS-PAGE and transfer to a PVDF membrane.
- Probe membranes with primary antibodies against key UPR proteins, followed by species-appropriate HRP-conjugated secondary antibodies.
- Key antibodies for UPR assessment include:
- Phospho-PERK and Total PERK
- Phospho-eIF2α and Total eIF2α
- ATF4
- CHOP
- XBP1s (requires specific antibodies that distinguish the spliced form)
- Use chemiluminescence for detection and normalize phospho-protein levels to total protein or a loading control (e.g., Actin).
Experimental Workflow for ER Stress Research: A standard methodology for inducing and analyzing the unfolded protein response in vitro.
Transcriptomic Analysis of ER Stress
For a systems-level view, RNA sequencing (RNA-Seq) provides an unbiased profile of the transcriptional response to ER stress.
Protocol: Transcriptomic Profiling of Cell-Type-Specific UPR [9]
Cell Treatment and RNA Preparation: Treat a panel of relevant cell lines (e.g., endocrine cells α, β, δ) with thapsigargin or vehicle for 6 h and 24 h. Isolve high-quality total RNA with an RNA Integrity Number (RIN) > 9.0.
Library Preparation and Sequencing: Prepare sequencing libraries using a standardized kit (e.g., poly-A selection). Sequence on an Illumina platform to a depth of at least 30 million reads per sample.
Bioinformatic Analysis:
- Align raw sequencing reads to the reference genome using a splice-aware aligner (e.g., STAR).
- Quantify transcript abundances and perform differential gene expression analysis using software packages (e.g., DESeq2).
- Identify shared and cell-type-specific transcriptional signatures by comparing differentially expressed genes across the different cell lines.
- Perform pathway enrichment analysis (e.g., Gene Ontology, KEGG) on gene sets upregulated by thapsigargin to identify biological processes most affected by ER stress.
The maintenance of ER homeostasis is a critical determinant of cellular health, with its disruption leading to ER stress and the activation of the UPR. The UPR's dual nature as both an adaptive and pro-apoptotic pathway highlights its importance in cell fate decisions. The experimental frameworks and reagents outlined herein provide a foundation for investigating this complex signaling network. A deep understanding of ER stress mechanisms continues to be essential for unraveling the pathophysiology of diverse diseases and for developing novel therapeutic strategies aimed at modulating the UPR.
The endoplasmic reticulum (ER) serves as a critical organelle for the synthesis, folding, and post-translational modification of secretory and transmembrane proteins, representing approximately one-third of the eukaryotic proteome [10] [11]. The environment within the ER is specialized for protein folding, featuring an oxidizing redox potential, high calcium concentration, and abundant chaperone proteins that assist in proper protein maturation [11]. Cellular stress induced by the abnormal accumulation of unfolded or misfolded proteins disrupts ER homeostasis, leading to a condition termed "ER stress" [10]. This stress arises from various pathological conditions including glucose or energy deprivation, calcium depletion, redox imbalance, hypoxia, genetic mutations, and increased protein synthesis demands [2] [11].
To counteract ER stress and restore protein homeostasis (proteostasis), eukaryotic cells have evolved an integrated signaling network known as the unfolded protein response (UPR) [10] [3]. The UPR operates through three major ER-transmembrane sensors: PERK (PKR-like ER kinase), IRE1α (inositol-requiring enzyme 1α), and ATF6 (activating transcription factor 6) [10] [12]. These sensors monitor protein folding status within the ER lumen and transmit information to the nucleus and cytosol, initiating adaptive programs that readjust the ER's protein-folding capacity [10]. Under physiological conditions, these sensors are maintained in an inactive state through association with the ER chaperone BiP (binding immunoglobulin protein, also known as GRP78) [13] [14]. During ER stress, BiP dissociates from the sensors to bind misfolded proteins, leading to sensor activation and UPR initiation [14] [15]. The primary goal of the UPR is to restore ER homeostasis through transcriptional and translational reprogramming that enhances protein folding, degradation of misfolded proteins, and reduction of incoming protein load [10] [3]. However, under severe or prolonged stress conditions, the UPR switches from pro-survival to pro-apoptotic signaling, eliminating severely damaged cells [12] [11].
Molecular Mechanisms of the Three UPR Sensors
PERK-eIF2α Signaling Pathway
PERK is a type I ER transmembrane protein that possesses serine/threonine kinase activity in its cytosolic domain [10] [12]. Upon ER stress, PERK undergoes oligomerization and trans-autophosphorylation, leading to its activation [10] [16]. The primary substrate of PERK is the α-subunit of eukaryotic translation initiation factor 2 (eIF2α), which PERK phosphorylates at serine 51 [10] [14]. This phosphorylation event represents a rapid adaptive response to ER stress by globally attenuating protein translation, thereby reducing the influx of newly synthesized proteins into the already stressed ER [10] [11].
Despite this global translational attenuation, phosphorylated eIF2α (p-eIF2α) selectively promotes the translation of specific mRNAs containing upstream open reading frames in their 5' untranslated regions [10]. One crucial transcription factor translated under these conditions is ATF4 (activating transcription factor 4), which activates genes involved in antioxidant response, amino acid metabolism, and apoptosis [10] [3]. ATF4 upregulates the pro-apoptotic transcription factor CHOP (C/EBP homologous protein, also known as GADD153) during prolonged ER stress [14] [3]. CHOP, in turn, promotes the expression of GADD34 (growth arrest and DNA damage-inducible protein 34), which forms a complex with protein phosphatase 1 to dephosphorylate eIF2α, restoring protein synthesis in a negative feedback loop [10] [3]. This PERK-eIF2α-ATF4-CHOP axis plays a critical role in determining cell fate decisions during ER stress, promoting survival under moderate stress but triggering apoptosis during irreversible damage [12] [3].
IRE1α-XBP1 Signaling Pathway
IRE1α is the most evolutionarily conserved UPR sensor, featuring both serine/threonine kinase and endoribonuclease activities in its cytosolic domain [10] [11]. Upon ER stress, IRE1α oligomerizes and autophosphorylates, activating its RNase domain [10] [12]. The primary substrate of IRE1α's RNase activity is XBP1 (X-box binding protein 1) mRNA [10]. IRE1α excises a 26-nucleotide intron from XBP1 mRNA through unconventional cytosolic splicing, resulting in a frameshift that produces the potent transcription factor XBP1s (spliced XBP1) [10] [16].
XBP1s translocates to the nucleus and activates genes encoding proteins involved in ER biogenesis, ER-associated degradation (ERAD), protein folding, and quality control [10] [12]. This transcriptional program expands the ER's protein-folding capacity and enhances its ability to degrade misfolded proteins [11]. Beyond XBP1 splicing, IRE1α also cleaves select mRNAs and pre-microRNAs through a process termed RIDD (regulated IRE1-dependent decay), which reduces the protein-folding load on the ER under stress conditions [10] [14]. During prolonged ER stress, IRE1α recruits TRAF2 (TNF receptor-associated factor 2), leading to activation of ASK1 (apoptosis signal-regulating kinase 1) and JNK (c-Jun N-terminal kinase), thereby promoting apoptotic signaling [12] [11]. Recent studies have revealed cross-talk between UPR branches, demonstrating that IRE1α-XBP1s signaling helps sustain PERK expression during chronic ER stress [16].
ATF6 Proteolytic Activation Pathway
ATF6 is a type II ER transmembrane protein that functions as a transcription factor [12]. Under normal conditions, ATF6 is retained in the ER through binding with BiP [12] [3]. During ER stress, ATF6 dissociates from BiP and translocates to the Golgi apparatus, where it undergoes sequential proteolytic cleavage by site-1 protease (S1P) and site-2 protease (S2P) [10] [12]. This regulated intramembrane proteolysis releases the cytosolic N-terminal domain of ATF6 (ATF6f), which translocates to the nucleus and functions as a potent transcription factor [3].
ATF6f activates the expression of genes encoding ER chaperones (including BiP), folding enzymes, and components of the ERAD pathway [12] [3]. The ATF6 pathway works cooperatively with the IRE1α-XBP1 pathway to enhance ER quality control mechanisms and restore proteostasis [10]. Mammalian cells express two ATF6 isoforms (ATF6α and ATF6β), with ATF6α exhibiting higher transcriptional activity [12] [14]. While single-knockout mice for either isoform are viable, double-knockout mice are embryonic lethal, indicating functional overlap during development [12]. Tissue-specific homologs of ATF6, including CREBH in hepatocytes and OASIS in astrocytes, have also been identified, suggesting specialized UPR regulation in different cell types [12].
Figure 1: Integrated UPR Signaling Pathways. The three ER stress sensors (PERK, IRE1α, and ATF6) are activated upon BiP dissociation during ER stress. Each sensor initiates distinct signaling cascades that collectively work to restore ER homeostasis through translational control, transcriptional regulation, and ER-associated degradation. Under prolonged stress, these pathways transition to promote apoptosis.
Comparative Analysis of UPR Signaling Branches
Table 1: Key Characteristics of the Three Major UPR Sensors
| Feature | PERK | IRE1α | ATF6 |
|---|---|---|---|
| Domain Structure | Type I transmembrane protein with kinase domain | Type I transmembrane protein with kinase and RNase domains | Type II transmembrane protein with bZIP transcription factor domain |
| Activation Mechanism | Oligomerization & trans-autophosphorylation | Oligomerization & trans-autophosphorylation | Transport to Golgi & proteolytic cleavage |
| Primary Signaling Output | eIF2α phosphorylation at Ser51 | XBP1 mRNA splicing | Release of cytosolic transcription factor domain (ATF6f) |
| Key Downstream Effectors | ATF4, CHOP, GADD34 | XBP1s, RIDD targets | ER chaperones, XBP1 |
| Primary Functions | Translational attenuation, antioxidant response, apoptosis regulation | ER biogenesis, quality control, ERAD, apoptosis regulation | ER chaperone induction, ERAD component synthesis |
| Physiological Roles | Metabolic adaptation, cell fate decisions | Plasma cell differentiation, professional secretory cell function | Development, tissue-specific UPR regulation |
| Knockout Phenotype | Postnatal lethality (pancreatic dysfunction) | Embryonic lethality (placental defects) | Viable (embryonic lethal in double ATF6α/β knockout) |
Table 2: UPR-Mediated Cell Fate Decisions Under Varying Stress Conditions
| Stress Condition | PERK Pathway | IRE1α Pathway | ATF6 Pathway | Overall Cell Fate |
|---|---|---|---|---|
| Acute/Mild Stress | Transient eIF2α phosphorylation, ATF4-mediated adaptation | XBP1 splicing enhances folding capacity & ERAD | Chaperone induction promotes protein refolding | Survival & homeostasis restoration |
| Chronic/Severe Stress | Sustained CHOP expression, GADD34-mediated feedback | RIDD activation, TRAF2-ASK1-JNK apoptosis signaling | Limited protective capacity | Apoptosis & cell elimination |
| Nutrient Deprivation | Amino acid metabolism reprogramming via ATF4 | Selective mRNA decay to conserve resources | Standard activation | Metabolic adaptation & survival |
| Hypoxia | Angiogenesis signaling via HIF-α stabilization | Limited contribution | Limited contribution | Angiogenesis & metabolic adaptation |
| Oxidative Stress | Antioxidant gene expression via ATF4 | Inflammatory signaling modulation | Standard activation | Redox homeostasis restoration |
Research Methodologies for UPR Investigation
Experimental Models for ER Stress Induction
The study of UPR signaling relies on well-established experimental approaches for inducing and monitoring ER stress. Chemical inducers that disrupt specific ER functions are commonly used:
- Tunicamycin: Inhibits N-linked glycosylation, causing accumulation of unglycosylated misfolded proteins [2].
- Thapsigargin: Inhibits the SERCA calcium pump, depleting ER calcium stores and disrupting calcium-dependent folding [2].
- Brefeldin A: Disrupts protein transport from ER to Golgi, causing protein accumulation in the ER [16].
Genetic models with knockout or knockdown of specific UPR components provide essential insights into their physiological functions. For example, IRE1α knockout causes embryonic lethality due to placental defects, while PERK knockout leads to postnatal lethality with pancreatic dysfunction [12]. Cell lines stably expressing reporter constructs (e.g., XBP1-GFP splice reporters) enable real-time monitoring of UPR activation dynamics [15].
Analytical Techniques for Monitoring UPR Activation
Table 3: Key Methodologies for UPR Pathway Analysis
| Technique | Application | Key Readouts | Experimental Considerations |
|---|---|---|---|
| Western Blot | Protein expression & phosphorylation | p-eIF2α, ATF4, CHOP, XBP1s, processed ATF6 | Requires specific antibodies; phosphorylation-specific antibodies needed |
| Quantitative PCR | Gene expression analysis | BiP, CHOP, XBP1s target genes, ERdj4 | Distinguish XBP1u vs XBP1s via restriction digest or specific primers |
| Blue Native PAGE | Monitor oligomerization states of UPR sensors | PERK & IRE1α complex formation | Maintains native protein complexes; assesses activation status [13] |
| Chromatin Immunoprecipitation (ChIP) | Transcription factor binding studies | XBP1s, ATF4, ATF6f genomic targets | Identifies direct vs indirect transcriptional targets [16] |
| Immunofluorescence/Confocal Microscopy | Subcellular localization | ATF6 Golgi translocation, XBP1s nuclear localization | Visualizes dynamic protein trafficking |
| XBP1 Splicing Assay | IRE1α RNase activity | XBP1 mRNA splicing pattern | RT-PCR with primers flanking splice site; PstI restriction digest distinguishes products |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for UPR Research
| Reagent/Category | Specific Examples | Function/Application | Mechanism of Action |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin, Thapsigargin, Brefeldin A | Experimental ER stress induction | Disrupt glycosylation, calcium homeostasis, or ER-Golgi transport [2] [16] |
| IRE1α Inhibitors | 4μ8C, MKC8866, KIRA6, STF-083010 | Specific inhibition of IRE1α RNase activity | Allosteric inhibition or competitive binding to RNase domain [2] [16] |
| PERK Inhibitors | GSK2606414, AMGEN 44 | Selective PERK kinase inhibition | ATP-competitive kinase inhibitors blocking eIF2α phosphorylation [16] [17] |
| Genetic Manipulation Tools | siRNA, CRISPR/Cas9 knockout, Inducible expression systems | Gene-specific knockdown/knockout or overexpression | Enables functional studies of specific UPR components [16] |
| Reporter Cell Lines | XBP1-GFP splice reporters, ERdj4-GFP, CHOP-GFP | Real-time monitoring of UPR activation dynamics | Links UPR element activity to fluorescent output for live imaging [15] |
| Activity-Specific Antibodies | p-eIF2α, XBP1s-specific, phospho-IRE1α | Detection of activated UPR components | Distinguishes active vs inactive forms of UPR signaling molecules |
Figure 2: Experimental Workflow for UPR Research. A systematic approach to investigating UPR signaling involves sequential phases of stress induction, pathway analysis, targeted manipulation, and data integration to build comprehensive models of ER stress responses.
Pathophysiological Implications and Therapeutic Targeting
The UPR plays critical roles in various pathological conditions, including metabolic diseases, neurodegeneration, cancer, and inflammatory disorders [10] [2] [14]. In cancer, tumor cells exploit UPR pathways to survive in the hostile tumor microenvironment characterized by hypoxia, nutrient deprivation, and oxidative stress [14] [17]. Each UPR branch contributes distinctly to tumor progression: PERK supports angiogenesis and metabolic adaptation; IRE1α-XBP1 signaling facilitates metastasis and inflammation; and ATF6 promotes resistance to apoptosis [17]. Cancer cells often display elevated basal UPR activity, making them particularly dependent on these pathways for survival [14].
Therapeutic strategies targeting UPR components are currently under development, particularly for oncology applications [17]. Small-molecule inhibitors of IRE1α RNase activity (e.g., 4μ8C, MKC8866) and PERK kinase inhibitors (e.g., GSK2606414) have shown promising results in preclinical models [16] [17]. Natural compounds like epigallocatechin gallate (EGCG) that target GRP78/BiP also demonstrate therapeutic potential [17]. Combination therapies linking UPR modulation with conventional chemotherapy or immunotherapy represent an emerging approach to overcome treatment resistance [17].
In immune cells, the UPR regulates development, activation, and effector functions [2]. XBP1 is essential for plasma cell differentiation and antibody production, while IRE1α signaling in macrophages promotes polarization toward pro-inflammatory phenotypes in metabolic diseases [2]. Neurodegenerative diseases including Alzheimer's, Parkinson's, and various retinal disorders involve chronic ER stress and UPR dysregulation [3]. Therapeutic approaches aimed at modulating UPR activation, such as chemical chaperones that facilitate protein folding, are being explored for these conditions [3].
The integral role of UPR signaling in human pathophysiology, combined with the development of specific pharmacological modulators, positions this cellular stress response system as a promising therapeutic target for multiple disease conditions. Future research will likely focus on tissue-specific UPR regulation and contextual activation patterns to develop more precise interventions with reduced off-target effects.
The endoplasmic reticulum (ER) serves as a critical organelle for protein folding, lipid synthesis, and calcium storage in eukaryotic cells. The disruption of its functional integrity leads to the accumulation of unfolded or misfolded proteins, a condition termed ER stress [3]. To mitigate this stress, cells activate a sophisticated signaling network known as the unfolded protein response (UPR) [18] [19]. The UPR is primarily mediated by three ER-transmembrane sensors: PERK (PKR-like ER kinase), IRE1 (Inositol-requiring enzyme 1), and ATF6 (Activating Transcription Factor 6) [19] [3]. Among these, the PERK branch constitutes a pivotal axis that orchestrates a rapid translational control program and, ultimately, determines cellular fate. The PERK-eIF2α-ATF4-CHOP pathway exemplifies the fundamental dichotomy of the UPR: it initially drives pro-adaptive responses to restore cellular homeostasis but can pivot to activate pro-apoptotic signaling under conditions of severe or prolonged stress [18] [20]. This whitepaper delineates the molecular mechanics, regulatory inputs, and functional outcomes of this critical signaling axis, framing it within the broader context of UPR research and its profound implications for human disease, particularly cancer.
Molecular Architecture of the PERK-eIF2α-ATF4-CHOP Signaling Axis
The PERK-mediated pathway is a sequential signaling cascade that translates the initial signal of ER protein misfolding into a definitive transcriptional and fate outcome.
Core Signaling Cascade
- PERK Activation and eIF2α Phosphorylation: Under homeostatic conditions, PERK is maintained in an inactive monomeric state through its association with the chaperone BiP (GRP78). The accumulation of unfolded proteins within the ER lumen triggers BiP dissociation, prompting PERK dimerization and trans-autophosphorylation [3] [20]. The activated PERK kinase then phosphorylates the alpha subunit of the eukaryotic translation initiation factor 2 (eIF2α) at serine 51. This phosphorylation event dramatically inhibits global protein synthesis by preventing the recycling of eIF2, thereby reducing the incoming protein load on the stressed ER [18] [3].
- Preferential Translation of ATF4: While phospho-eIF2α (p-eIF2α) attenuates most cap-dependent translation, it paradoxically promotes the selective translation of specific mRNAs, most notably that of Activating Transcription Factor 4 (ATF4) [18] [3]. This occurs through upstream open reading frames (uORFs) in the ATF4 mRNA that are bypassed under these stressful conditions.
- CHOP Induction and Fate Determination: ATF4 functions as a master transcriptional regulator of the integrated stress response, upregulating genes involved in amino acid metabolism, antioxidant responses, and autophagy. A key transcriptional target of ATF4 is C/EBP-homologous protein (CHOP, also known as GADD153) [18] [20]. Under transient ER stress, the PERK-ATF4-CHOP axis promotes adaptive recovery. However, persistent stress leads to sustained CHOP expression, which drives the expression of pro-apoptotic genes, including those encoding death receptor 5 (DR5) and ER oxidase 1α (ERO1α), while simultaneously suppressing the anti-apoptotic Bcl-2 protein, thereby committing the cell to apoptosis [18] [3].
The following diagram illustrates the core logic and key components of this pathway:
Key Regulatory Mechanisms and Interacting Partners
The core pathway is subject to intricate regulatory feedback and crosstalk with other cellular processes. GADD34, a subunit of a phosphatase complex that is itself a transcriptional target of CHOP, dephosphorylates eIF2α to restore protein synthesis and facilitate recovery from transient stress [3]. However, under irremediable stress, this restoration of translation may paradoxically contribute to cell death by increasing the protein-folding burden. Furthermore, the PERK axis exhibits significant crosstalk with other UPR branches and with signaling pathways outside the ER. For instance, ATF4 can cooperate with the transcription factor XBP1s (generated by the IRE1 branch) to regulate a common set of target genes [20]. The pathway is also intimately linked with oxidative stress; CHOP-upregulated ERO1α can promote hyper-oxidation of the ER environment, further exacerbating stress and promoting cell death [3].
Experimental Dissection: Methodologies for Investigating the Pathway
A robust experimental framework is essential for dissecting the complex roles of the PERK-eIF2α-ATF4-CHOP axis. The table below summarizes key methodologies and reagents used to probe this pathway.
Table 1: Key Research Reagent Solutions for Investigating the PERK-eIF2α-ATF4-CHOP Axis
| Reagent / Assay | Function / Target | Experimental Application | Key Findings Enabled |
|---|---|---|---|
| Thapsigargin (Tg) | SERCA pump inhibitor; disrupts ER calcium homeostasis [19] | Induces ER stress and activates UPR sensors, including PERK. | Elucidated PERK activation kinetics and its role in translational control and apoptosis [18]. |
| Tunicamycin (Tm) | Inhibits N-linked protein glycosylation [19] | Triggers ER stress by causing accumulation of unfolded glycoproteins. | Demonstrated the link between glycosylation defects, PERK activation, and CHOP-mediated apoptosis [18]. |
| GSK2606414 | Potent and selective PERK inhibitor [17] [21] | Inhibits PERK kinase activity to block the entire downstream axis. | Validated the PERK pathway's role in tumor cell survival and as a therapeutic target in cancer [17]. |
| ISRIB | Integrated Stress Response inhibitor; reverses eIF2α phosphorylation effects [21] | Rescues translational attenuation downstream of p-eIF2α. | Distinguished effects specific to eIF2α phosphorylation from other PERK functions [21]. |
| Genetic Knockout MEFs (e.g., Perk⁻⁻, Atf4⁻⁻) [21] | Deletion of specific pathway components. | Provides definitive evidence for a gene's function in stress-induced signaling and fate decisions. | Confirmed the essential role of PERK and ATF4 in mediating ER-stress-induced cell death and ferroptosis [21]. |
Detailed Protocol: Assessing Pathway Activation and Functional Consequences
A standard workflow for investigating this axis involves inducing ER stress, followed by monitoring pathway components and assessing the ultimate cellular outcome.
Step 1: Induction of ER Stress. Treat cells with well-characterized ER stress inducers. Common protocols use Thapsigargin (250 nM - 1 μM) or Tunicamycin (1 - 5 μg/mL) for 1 to 24 hours, depending on the desired response strength and time point [19]. Include a vehicle control (e.g., DMSO) for comparison.
Step 2: Monitoring Pathway Activation.
- Western Blot Analysis: Resolve cell lysates by SDS-PAGE and probe for key markers.
- Quantitative RT-PCR: Measure mRNA levels of ATF4 and CHOP (DDIT3) to assess transcriptional upregulation [19].
Step 3: Determining Functional Outcomes.
- Cell Viability Assays: Use MTT, Annexin V/PI staining, or Caspase-3/7 activity assays to quantify apoptosis following sustained ER stress (e.g., 24-48 hours of treatment) [23].
- Translational Profiling: Employ the SUnSET (SUrface SEnsing of Translation) assay, which involves puromycin incorporation into nascent polypeptides, to directly measure global protein synthesis rates and their inhibition by p-eIF2α [23].
The following workflow diagram outlines the key steps of this experimental process:
Functional Outcomes and Pathological Significance
The PERK-eIF2α-ATF4-CHOP axis is a central determinant of cell fate, and its dysregulation underpins a spectrum of human diseases.
The Dichotomy of Cell Fate Decisions
The functional outcome of pathway activation is context-dependent, hinging on the duration and intensity of the stress signal.
- Pro-Adaptive Response: In the face of transient stress, the pathway promotes survival. The rapid inhibition of global translation reduces the ER's protein-folding load. Concurrently, ATF4-driven transcription upregulates genes involved in amino acid metabolism, antioxidant responses, and autophagy, collectively working to restore proteostasis [3] [20].
- Pro-Apoptotic Switch: Under severe, prolonged, or irremediable ER stress, the sustained induction of CHOP orchestrates a shift toward apoptosis. CHOP downregulates the anti-apoptotic protein Bcl-2 and upregulates pro-apoptotic proteins like BIM and DR5. Furthermore, CHOP-induced ERO1α can lead to oxidative stress, and the GADD34-mediated restoration of protein synthesis can exacerbate the ER protein load, collectively triggering mitochondrial apoptosis [18] [3].
Role in Tumor Biology and Therapy Resistance
The tumor microenvironment, characterized by hypoxia, nutrient deprivation, and acidosis, imposes chronic ER stress on cancer cells. Consequently, the PERK-eIF2α-ATF4-CHOP axis is frequently co-opted to support oncogenic adaptation [18] [17]. It promotes angiogenesis through ATF4-mediated induction of VEGF and other pro-angiogenic factors. It facilitates metastatic dissemination by aiding cancer cells in adapting to detachment-induced stress (anoikis resistance). Furthermore, this pathway is a key mediator of resistance to conventional chemotherapy and radiotherapy, as these treatments further exacerbate ER stress, and cancer cells rely on the adaptive UPR to survive [17]. This creates a therapeutic vulnerability; targeting the PERK pathway can sensitize tumors to standard treatments.
Table 2: Quantitative Findings Linking the PERK Axis to Disease Pathogenesis
| Pathological Context | Key Molecular Finding | Functional/Clinical Correlation | Source |
|---|---|---|---|
| Breast Cancer | ERS inducers (Thapsigargin, Brefeldin A) reduce ERα protein & mRNA via PERK/eIF2α/ATF4. | ATF4 binds ESR1 promoter, suppressing its activity. Selective PERK activation inhibits tumor growth in vivo. [22] | Laboratory Study |
| Head and Neck Cancer | Tumors with hypoxic conditions are more resistant to radiation therapy. | Hypoxia-induced PERK/eIF2α/ATF4 signaling promotes pro-adaptive survival, contributing to poor clinical outcomes. [18] | Clinical Observation |
| Diet-Induced Obesity & Insulin Resistance | Saturated fatty acids engage PERK-ATF4 axis in macrophages, inducing pro-inflammatory IL-6. | CHOP expression in adipocytes drives pro-inflammatory M1 macrophage polarization, resulting in insulin resistance. [19] | Preclinical Model |
| Acetaminophen (APAP)-Induced Hepatotoxicity | Inhibition of eIF2α-ATF4 or ferroptosis protects against APAP-induced liver damage. | The PERK-eIF2α-ATF4 axis was identified as a crucial mediator of ferroptotic cell death in this liver injury model. [21] | Preclinical Model |
Therapeutic Targeting and Future Directions
The critical role of the PERK pathway in diseases like cancer has galvanized efforts to develop targeted therapeutics. Two primary strategies have emerged: inhibition of the pro-survival output to sensitize cells to stress, and hyperactivation of the pathway to push cells toward apoptosis.
Inhibitory Strategies: Small-molecule inhibitors of PERK (e.g., GSK2606414) have shown efficacy in preclinical cancer models, particularly in combination with other chemotherapeutic agents that induce ER stress, by blocking the tumor's adaptive response [17]. Similarly, inhibitors of the IRE1 RNase domain are also in development.
Activator Strategies: An alternative approach is the selective activation of the PERK branch or the inhibition of feedback regulators like GADD34 to push the stressed cell over the apoptotic threshold. This leverages the inherent duality of the pathway for therapeutic gain.
A major challenge in this field is the context-dependent nature of the pathway's output. Factors such as tumor type, genetic background, and the dynamics of the tumor microenvironment can influence whether PERK inhibition or activation will be beneficial [17] [20]. Future research must focus on delineating these contextual determinants to enable precision targeting of the UPR in human disease. The integration of UPR modulators with emerging modalities like immunotherapy, given the role of ER stress in shaping the tumor immune microenvironment, represents a particularly promising frontier [17].
The endoplasmic reticulum (ER) is the largest organelle in the mammalian cell and serves as the primary site for the synthesis, folding, and modification of secretory and membrane proteins, as well as lipid biosynthesis and calcium storage [2] [3] [24]. The accumulation of unfolded or misfolded proteins in the ER lumen causes ER stress, triggering a highly conserved adaptive mechanism known as the unfolded protein response (UPR) [2] [3]. The UPR aims to restore ER homeostasis by reducing global protein synthesis, enhancing the ER's protein-folding capacity, and degrading misfolded proteins. However, under severe or prolonged stress, the UPR can initiate apoptotic cell death [3] [24].
In mammalian cells, the UPR is initiated by three ER-transmembrane sensors: IRE1α (inositol-requiring enzyme 1α), PERK (PKR-like ER kinase), and ATF6 (activating transcription factor 6) [2] [3]. Among these, IRE1α is the most ancient and evolutionarily conserved branch [25] [24]. It is a type I transmembrane protein featuring an ER-luminal stress-sensing domain and a cytosolic domain possessing both kinase and endoribonuclease activities [26] [25]. Activation of IRE1α's RNase function orchestrates a complex, multi-layered response through two distinct outputs: the non-conventional splicing of X-box binding protein 1 (XBP1) mRNA and the regulated degradation of a subset of cellular mRNAs, a process known as Regulated IRE1α-Dependent Decay (RIDD) [26] [27] [25]. This whitepaper provides an in-depth technical guide to the IRE1α-XBP1 pathway and RIDD, framing them within the broader context of UPR research and their implications for therapeutic drug development.
Molecular Mechanisms of IRE1α Signaling
IRE1α Activation and Oligomerization
Under homeostatic conditions, IRE1α is maintained in an inactive monomeric state through its association with the ER chaperone BiP (Binding Immunoglobulin Protein, also known as GRP78) [2] [24]. An accumulation of unfolded proteins creates a demand for chaperone function, drawing BiP away from IRE1α's luminal domain. This dissociation allows IRE1α to dimerize and oligomerize [26] [27]. Oligomerization facilitates trans-autophosphorylation of the kinase domain, which induces a conformational change that activates the RNase domain [27] [25]. The core mechanism of IRE1α activation is illustrated in the following pathway diagram:
The Dual Outputs of IRE1α RNase Activity
The activated RNase domain of IRE1α executes two critical functions, which can be modulated by its oligomerization state:
XBP1 Splicing: IRE1α cleaves the XBP1 mRNA at two specific stem-loop structures, removing a 26-nucleotide intron [26] [3]. The excised fragments are then ligated by the tRNA ligase RTCB, resulting in a frameshift and the production of the spliced mRNA (XBP1s) [26]. This mRNA is translated into the potent transcription factor XBP1s, which translocates to the nucleus and drives the expression of genes involved in ER biogenesis, protein folding, secretion, and ER-associated degradation (ERAD) [26] [28] [3].
Regulated IRE1α-Dependent Decay (RIDD): In parallel, IRE1α cleaves other mRNAs, leading to their degradation [26] [27] [25]. Initially, mammalian RIDD was thought to target only mRNAs possessing a stem-loop structure with a consensus loop sequence similar to that in XBP1 mRNA (CNG|CAGN) [27]. However, recent research has identified a more promiscuous activity, termed RIDDLE (RIDD Lacking Endomotif), which targets mRNAs without this canonical motif [27]. The modality of cleavage is determined by the activation state of IRE1α: dimeric IRE1α preferentially performs endomotif-directed cleavage, while phospho-oligomeric IRE1α is required for endomotif-independent RIDDLE [27].
The following diagram delineates the experimental workflow and logical relationships used to study these two RNase outputs:
Table 1: Core Functions of IRE1α RNase Outputs
| Function | Mechanism | Primary Sequence Motif | IRE1α State | Biological Role |
|---|---|---|---|---|
| XBP1 Splicing | Cleaves XBP1u mRNA, causing a frameshift to produce XBP1s [26] [3] | CNG|CAGN (within a stem-loop) [27] | Dimer/Oligomer | Pro-survival: Produces XBP1s transcription factor to enhance ER folding capacity [26] [25] |
| RIDD | Cleaves and degrades select cellular mRNAs [26] [27] | CNG|CAGN (canonical) [27] | Dimer | Pro-survival & Pro-death: Reduces ER protein-folding load; can trigger apoptosis under prolonged stress [26] [25] |
| RIDDLE | Cleaves mRNAs lacking the canonical endomotif [27] | Endomotif-independent [27] | Phospho-oligomer | Context-dependent: Contributes to mRNA decay under severe stress [27] |
Quantitative Data in IRE1α Research
Lipidomic and Transcriptomic Profiling Data
Integrative omics approaches have been pivotal in uncovering the role of IRE1α in lipid metabolism. A study inhibiting IRE1α's RNase with MKC8866 in triple-negative breast cancer (TNBC) cells (MDA-MB-231) revealed significant, time-dependent alterations in lipid profiles and gene expression [26].
Table 2: Lipidomic Changes in MDA-MB-231 Cells Upon IRE1α Inhibition (MKC8866 Treatment) [26]
| Lipid Class | Change at 48-72h | Specific Alterations | Proposed Interpretation |
|---|---|---|---|
| Triacylglycerols (TAGs) | ↑ Increased | Preferential increase in mono- and polyunsaturated long-chain species; decrease in saturated TAGs [26] | Shift in lipid metabolism favoring TAG synthesis and storage in lipid droplets [26] |
| Diacylglycerols (DAGs) | ↓ Decreased | General decrease across species [26] | Precursor consumption for elevated TAG synthesis [26] |
| Free Fatty Acids (FAs) | ↓ Decreased | Reduction in 18-23 carbon MUFAs and PUFAs [26] | Incorporation into newly synthesized TAGs [26] |
| Phosphatidylcholines (PCs) | ↓ Decreased | Reduced levels of polyunsaturated and elongated species [26] | Membrane phospholipid remodeling to support neutral lipid storage [26] |
| Ceramides | ↓ Decreased | General reduction [26] | Broader impact on lipid metabolism beyond neutral lipids [26] |
Concurrent transcriptomic analysis identified 395-883 differentially expressed genes (DEGs) upon IRE1α inhibition, with a significant number involved in lipid metabolism [26]. Key upregulated genes included:
- DGAT2: Encoding diacylglycerol O-acyltransferase 2, the rate-limiting enzyme in TAG biosynthesis [26].
- SREBF1: Encoding a master transcription factor regulating sterol and fatty acid synthesis.
- LIPE: Encoding hormone-sensitive lipase, involved in lipid droplet breakdown [26].
Experimentally Identified RIDD Substrates
The scope of RIDD targets has been expanded through modern sequencing techniques. A study integrating total RNA-seq and GRO-seq (Global Run-On Sequencing) in ER-stressed human cells identified 54 mRNAs as potential RIDD substrates, defined by an IRE1α-dependent decrease in abundance without a corresponding decline in transcription [27].
Table 3: Experimentally Validated RIDD Target mRNAs and Their Functions [26] [27]
| Gene Name | Protein Function | Experimental Context | Validation Method |
|---|---|---|---|
| DGAT2 | Rate-limiting enzyme in triacylglycerol synthesis [26] | TNBC (MDA-MB-231); IRE1α inhibition/activation [26] | RNA-seq, RT-qPCR, Actinomycin D assay [26] |
| CD59 | Glycosylphosphatidylinositol (GPI)-anchored complement regulatory protein [27] | MDA-MB-231, U2OS, HCT116 cells; Tg/Tm treatment [27] | Integrated RNA-seq/GRO-seq, RT-qPCR [27] |
| COL6A1 | Extracellular matrix protein (Collagen type VI alpha 1 chain) | MDA-MB-231 cells; Tg treatment [27] | Integrated RNA-seq/GRO-seq [27] |
| TGOLN2 | Intracellular protein trafficking (Trans-Golgi network protein 2) | MDA-MB-231 cells; Tg treatment [27] | Integrated RNA-seq/GRO-seq, RT-qPCR [27] |
| BLOS1 | Subunit of BLOC-1 complex involved in autophagy and lysosome biogenesis [27] | MDA-MB-231 cells; Tg treatment [27] | Integrated RNA-seq/GRO-seq [27] |
Detailed Experimental Protocols
This section outlines key methodologies used to dissect IRE1α-XBP1 signaling and RIDD activity, providing a reference for researchers aiming to replicate or adapt these approaches.
Protocol: Assessing IRE1α RNase Activity via XBP1 Splicing and RIDD
Objective: To evaluate IRE1α activation by measuring the splicing of XBP1 mRNA and the decay of a known RIDD target (e.g., DGAT2).
Key Reagents:
- ER Stress Inducers: Tunicamycin (Tm, 1-5 µg/mL), Thapsigargin (Tg, 0.1-1 µM) [26] [29].
- IRE1α RNase Inhibitors: MKC8866 (e.g., 50 µM) or STF-083010 (e.g., 50 µM) [26] [2].
- Cell Lines: MDA-MB-231 (TNBC), HepG2 (hepatoma), or MEFs (wild-type and IRE1α-/-) [26] [27] [29].
Procedure:
- Cell Treatment and Lysis: Seed cells and treat with stressors/inhibitors for predetermined time points (e.g., 3h, 8h, 24h). Extract total RNA using a commercial kit (e.g., TRIzol).
- cDNA Synthesis: Reverse transcribe 1 µg of total RNA using an M-MLV First-Strand cDNA Synthesis Kit with random hexamers or oligo(dT) primers.
- Quantitative PCR (qPCR):
- Perform qPCR using SYBR Green master mix.
- Design primers to distinguish XBP1u and XBP1s (amplicons spanning the splice junction) or to detect RIDD targets like DGAT2.
- Use housekeeping genes (e.g., GAPDH, ACTB) for normalization.
- Data Analysis:
- Calculate the XBP1 splicing ratio as: [XBP1s / (XBP1u + XBP1s)] × 100% [29].
- For RIDD targets, analyze relative mRNA expression using the 2^(-ΔΔCt) method, comparing treated samples to controls.
Protocol: Identifying Novel RIDD Substrates via Integrated RNA-seq and GRO-seq
Objective: To comprehensively identify direct RIDD targets by distinguishing transcriptional from post-transcriptional regulation.
Key Reagents:
- Nuclei Isolation Buffer: For GRO-seq sample preparation.
- GRO-seq Reagents: Bromouridine (BrU) triphosphate, anti-BrU antibodies, and reagents for nuclear run-on.
- RNA-seq Library Prep Kit: For standard total RNA sequencing.
Procedure [27]:
- Parallel Sample Preparation:
- For total RNA-seq, extract and purify total RNA from ER-stressed and control cells.
- For GRO-seq, isolate nuclei from parallel cell cultures. Perform the global nuclear run-on reaction with BrU-labeled nucleotides. Isolate the nascent, BrU-incorporated RNA using anti-BrU antibodies.
- Library Preparation and Sequencing: Prepare sequencing libraries from both total RNA and GRO-seq nascent RNA. Sequence on an appropriate platform (e.g., Illumina).
- Bioinformatic Analysis:
- Map sequencing reads to the reference genome.
- For total RNA-seq, quantify steady-state mRNA levels.
- For GRO-seq, quantify nascent transcription rates.
- Target Identification:
- Identify mRNAs where steady-state levels decrease significantly (e.g., fold change < -1.4) in an IRE1α-dependent manner (comparing WT vs. KO cells).
- Filter this list to retain only those mRNAs whose nascent transcription rates do not decrease correspondingly.
- The resulting mRNAs are high-confidence candidates for direct RIDD cleavage.
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for Investigating IRE1α-XBP1 Signaling and RIDD
| Reagent / Tool | Function / Target | Key Use Cases & Notes |
|---|---|---|
| Tunicamycin (Tm) | Inhibits N-linked glycosylation, inducing ER stress and IRE1α activation [2] [29] | Positive control for UPR induction; used at 1-5 µg/mL [26] [29]. |
| Thapsigargin (Tg) | Inhibits SERCA pump, disrupting ER calcium homeostasis and inducing ER stress [2] [29] | Positive control for UPR induction; potent and fast-acting; used at 0.1-1 µM [26] [27]. |
| MKC8866 | Selective inhibitor of IRE1α's RNase activity [26] | Functional studies to dissect RNase-dependent effects; used at ~50 µM [26]. |
| STF-083010 | Selective inhibitor of IRE1α's RNase activity [2] | Ameliorates insulin resistance in obesity models; inhibits RIDD [2]. |
| 4µ8C | Selective inhibitor of IRE1α's RNase activity [30] | Used to block IRE1α-driven IL-6 expression in melanoma studies [30]. |
| IRE1α-/- Cells | Genetic knockout of IRE1α | Essential control for confirming IRE1α-dependency of any observed phenotype [27]. |
| XBP1-/- Cells | Genetic knockout of XBP1 | Used to dissect XBP1s-dependent and -independent (e.g., RIDD) functions of IRE1α [26]. |
| Anti-XBP1s Antibody | Specifically detects the spliced, active form of XBP1 protein [30] | Critical for validating IRE1α pathway activation via immunoblotting or IHC [30]. |
| Anti-phospho-IRE1α Antibody | Detects phosphorylated (activated) IRE1α | Used to assess IRE1α activation via immunoblotting; shows mobility shift on SDS-PAGE [28]. |
Cross-Talk and Broader Research Context
The IRE1α-XBP1 pathway does not function in isolation. Significant cross-talk exists with other UPR branches and cellular processes. A key integrative mechanism is the PERK-ATF4 pathway-mediated upregulation of IRE1α expression. During ER stress, PERK phosphorylates eIF2α, leading to the selective translation of the transcription factor ATF4, which in turn binds to the IRE1α promoter and increases its transcription [29]. This creates a positive feedback loop, enhancing the cell's capacity to splice XBP1 and intensifying the IRE1α-mediated adaptive response [29].
Furthermore, IRE1α signaling plays critical, context-dependent roles in various physiological and pathological states beyond the basic UPR:
- Adipogenesis: The IRE1α-XBP1 pathway is indispensable for adipocyte differentiation. XBP1s is induced by the early adipogenic factor C/EBPβ and, in turn, directly activates the promoter of C/EBPα, a master regulator of adipogenesis [28].
- Cancer: In triple-negative breast cancer, IRE1α's RIDD activity targets DGAT2 mRNA to reprogram lipid metabolism, preventing TAG accumulation and protecting cells from metabolic stress [26]. In melanoma, the IRE1α-XBP1 pathway is constitutively active and promotes tumor progression by directly transactivating IL-6, which subsequently activates the JAK/STAT3 signaling pathway in an autocrine/paracrine manner [30].
- Immune Cell Function: IRE1α is a key regulator in immune cells such as B cells, macrophages, and dendritic cells. In macrophages, IRE1α activation by saturated fatty acids can promote NLRP3 inflammasome activation and IL-1β secretion, linking ER stress to inflammation and insulin resistance [2].
The IRE1α-XBP1 signaling axis and RIDD represent a sophisticated, multi-layered system central to the unfolded protein response. The precise balance between the pro-survival outputs of XBP1 splicing and the potentially dual-natured RIDD activity determines cellular fate in the face of proteotoxic stress. Advanced techniques like integrated transcriptomics and lipidomics, coupled with specific pharmacological and genetic tools, have revealed the profound influence of this pathway on diverse processes including lipid metabolism, immune regulation, and cancer progression. The intricate cross-talk with other stress response pathways and its context-dependent roles in disease highlight IRE1α as a compelling, albeit complex, target for therapeutic intervention. Future research will continue to unravel the molecular logic governing the choice between its adaptive and pro-death outputs, paving the way for novel treatments for cancer, metabolic diseases, and neurodegeneration.
ATF6 Proteolytic Activation and Transcriptional Regulation of ER Chaperones
The endoplasmic reticulum (ER) serves as a crucial cellular compartment for the synthesis, folding, and modification of secretory and transmembrane proteins. The accumulation of unfolded proteins within the ER lumen triggers a condition known as ER stress, which activates an evolutionarily conserved signaling network termed the unfolded protein response (UPR) [31]. The UPR aims to restore ER proteostasis through three primary branches, each regulated by distinct ER-resident transmembrane sensors: IRE1, PERK, and ATF6 [32]. This review focuses on the molecular mechanisms of ATF6 activation and its specialized role as a transcriptional regulator of ER chaperones and quality control factors.
Among the three UPR sensors, ATF6 functions as a master transcriptional regulator that enhances the ER's protein-folding capacity. While IRE1 and PERK primarily mediate translational control and ER expansion, ATF6 activation leads to the upregulation of genes encoding ER chaperones, folding enzymes, and components of ER-associated degradation (ERAD) [33]. Genome-wide analyses reveal that ATF6 controls a relatively small, selective set of target genes, with approximately 40% of these being ER quality control proteins, underscoring its specialized role in proteostasis maintenance [33]. The precise regulation of ATF6 proteolytic activation and its transcriptional network represents a critical adaptive mechanism in cellular stress responses, with implications for various disease pathologies including neurodegeneration, sensory disorders, and cancer.
Proteolytic Activation Mechanism of ATF6
Initial Structure and ER Retention
ATF6 is constitutively synthesized as a 90-kDa type II transmembrane protein (p90ATF6) embedded in the ER membrane. Its structure features an N-terminal cytosolic domain containing a bZIP transcription factor motif, a single transmembrane domain, and a C-terminal luminal portion that senses ER folding status [31] [34]. In unstressed conditions, ATF6 is retained in the ER through its association with the ER chaperone BiP/GRP78, which binds to the luminal domain and masks ER export signals, thereby preventing premature activation [35] [34].
ER Stress-Induced Activation and Golgi Translocation
Upon accumulation of unfolded proteins in the ER lumen, BiP dissociates from ATF6 to engage misfolded clients, unmasking the ER export signals on ATF6's luminal domain [35]. This triggers the selective packaging of ATF6 into COPII-coated vesicles for transport to the Golgi apparatus. The dissociation of BiP represents a crucial regulatory step that directly links the protein-folding status in the ER lumen to ATF6 activation signaling [34].
Regulated Intramembrane Proteolysis
Within the Golgi apparatus, ATF6 undergoes sequential proteolytic processing by two resident proteases. First, the Site-1 protease (S1P) cleaves ATF6 within its luminal domain, after which the Site-2 protease (S2P) cleaves within the transmembrane segment [35] [34]. This regulated intramembrane proteolysis (RIP) releases the soluble 50-kDa N-terminal fragment (p50ATF6) containing the complete bZIP transcription factor domain [31].
Nuclear Translocation and DNA Binding
The liberated p50ATF6 translocates to the nucleus where it functions as a potent transcription factor. Nuclear ATF6 binds to specific cis-acting elements in target gene promoters, predominantly the ER stress response element (ERSE) with a consensus sequence CCAAT-N₉-CCACG [31] [34]. ATF6 exhibits cooperative binding with the ubiquitous transcription factor NF-Y (also known as CBF), which recognizes the CCAAT portion of ERSE, while ATF6 binds the CCACG motif [31]. This cooperative interaction ensures precise transcriptional control of UPR target genes.
The following diagram illustrates the complete ATF6 activation pathway:
Figure 1: ATF6 Proteolytic Activation Pathway. During ER stress, ATF6 translocates from the ER to the Golgi where it undergoes sequential cleavage by S1P and S2P, releasing its transcription factor domain that travels to the nucleus to activate ER chaperone genes.
Transcriptional Regulation of ER Chaperones by ATF6
The ER Stress Response Element (ERSE)
The ER stress response element serves as the primary DNA regulatory sequence through which ATF6 controls target gene expression. The canonical ERSE has a tripartite structure with a consensus sequence of CCAAT-N₉-CCACG, where N₉ represents any nine nucleotides [31] [34]. The CCAAT motif is recognized and bound by the general transcription factor NF-Y (also known as CBF), while the CCACG motif provides the specific binding site for ATF6 [31]. This spatial arrangement, with precisely spaced motifs, enables cooperative binding between NF-Y and ATF6, creating a highly specific transcriptional complex that activates gene expression only during genuine ER stress conditions.
Research has demonstrated that ATF6 binding to the CCACG element is strictly dependent on NF-Y already being bound to the adjacent CCAAT sequence [31]. This molecular cooperation explains the stringent specificity of ATF6-mediated transcription in the UPR. Some target genes contain variant ERSE sequences, such as the ATTGG-N-CCACG motif found in the human Herp promoter, suggesting some flexibility in the exact configuration of ATF6-NF-Y interactions [34].
Key ER Chaperone Targets
ATF6 directly regulates numerous ER chaperones and folding factors essential for protein quality control. Major targets include:
- BiP/GRP78: The central ER Hsp70 chaperone that binds unfolded proteins and regulates all three UPR sensors [34]
- GRP94: An ER Hsp90 homolog involved in folding secreted proteins and integrins [31]
- Calreticulin: A major calcium-binding chaperone in the ER lumen [31]
- Protein disulfide isomerase (PDI): Catalyzes disulfide bond formation and isomerization [31]
- ERp72: A PDI-family oxidase involved in disulfide bond formation [31]
Beyond these classical ER chaperones, ATF6 also regulates transcription of other UPR components, including CHOP/GADD153 and XBP-1, creating interconnected feedback loops that modulate the overall stress response [31]. The diversity of ATF6 targets highlights its central role in enhancing multiple aspects of ER folding capacity during stress conditions.
Quantitative Analysis of ATF6 Target Genes
Table 1: Major ER Chaperone and Folding Factor Genes Regulated by ATF6
| Gene Target | Protein Function | ERSE Sequence in Promoter | Regulation by ATF6 |
|---|---|---|---|
| BiP/GRP78 | ER Hsp70 chaperone, master UPR regulator | CCAAT-N₉-CCACG | Direct activation [34] |
| GRP94 | ER Hsp90 chaperone, glycoprotein folding | CCAAT-N₉-CCACG | Direct activation [31] |
| Calreticulin | Calcium-binding chaperone | CCAAT-N₉-CCACG | Direct activation [31] |
| Protein Disulfide Isomerase (PDI) | Disulfide bond formation | CCAAT-N₉-CCACG | Direct activation [31] |
| FKBP13 | Peptidyl-prolyl cis-trans isomerase | Not specified in sources | Induction in ER stress [31] |
| ERp72 | Protein disulfide isomerase family member | Not specified in sources | Induction in ER stress [31] |
| CHOP/GADD153 | Pro-apoptotic transcription factor | CCAAT-N₉-CCACG | Direct activation [31] |
| XBP-1 | UPR transcription factor | CCAAT-N₉-CCACG | Direct activation [31] |
Genome-wide studies have revealed that ATF6 regulates a selective transcriptional program rather than broadly activating ER-related genes. Analysis of ATF6 target genes in mouse models demonstrated that only 30 out of 14,729 analyzable genes qualified as specific ATF6 targets, with approximately 40% of these encoding ER quality control proteins, 20% encoding other ER proteins, and the remainder having miscellaneous functions [33]. This selective regulation emphasizes ATF6's specialized role in maintaining ER proteostasis compared to the broader functions of IRE1 and PERK branches.
Experimental Analysis of ATF6 Activation
Key Methodologies and Reagents
The study of ATF6 proteolytic activation and function employs specialized experimental approaches that enable researchers to monitor its complex regulation. Key methodologies include luciferase reporter assays to measure ATF6 transcriptional activity, immunoblotting to detect the proteolytic conversion from p90 to p50 forms, immunofluorescence to visualize subcellular localization, and molecular techniques to assess DNA binding specificity [35] [34].
Table 2: Essential Research Reagents for ATF6 Investigation
| Research Tool | Application | Experimental Function |
|---|---|---|
| p5×ATF6-GL3 Reporter | Luciferase reporter assay | Measures ATF6 transcriptional activity via 5xATF6 binding sites [34] |
| GAL4-ATF6 Fusion System | Luciferase reporter assay | Assesses ATF6 activation using GAL4 DNA-binding domain [34] |
| ATF6 (373) Construct | Functional studies | Expresses constitutively active ATF6 fragment (amino acids 1-373) [36] |
| Site-1 Protease Inhibitor (PF-429242) | Pharmacological inhibition | Blocks ATF6 processing by inhibiting S1P [35] |
| Anti-ATF6 Antibodies | Immunodetection | Recognizes full-length (p90) and cleaved (p50) ATF6 forms [35] |
| ER Stress Inducers (Tunicamycin, Thapsigargin) | Experimental induction | Triggers ER stress and subsequent ATF6 activation [31] |
| BiP/GRP78 siRNA | Functional knockdown | Reduces BiP expression to study ATF6 regulation [35] |
Detailed Experimental Workflow
The following diagram outlines a comprehensive experimental approach for analyzing ATF6 proteolytic activation:
Figure 2: Experimental Workflow for Analyzing ATF6 Activation. Comprehensive approach combining protein-based techniques to monitor ATF6 processing and cellular localization with gene expression methods to assess its transcriptional activity.
Critical Experimental Considerations
When investigating ATF6 activation, several technical considerations are essential for accurate interpretation. First, the kinetics of ATF6 processing must be carefully timed, as proteolytic cleavage occurs within hours of stress induction, preceding the upregulation of target genes like BiP [31]. Second, researchers should account for the functional differences between ATF6 isoforms (ATF6α and ATF6β), with ATF6α serving as the primary transcriptional activator while ATF6β may have modulatory or inhibitory functions [34]. Third, the cell type-specific responses should be considered, as ATF6 activation dynamics may vary across different tissues and experimental systems.
For luciferase reporter assays, the p5×ATF6-GL3 construct containing five tandem copies of the ATF6 binding site provides a sensitive system for monitoring ATF6 transcriptional activity [34]. This approach can be complemented with the GAL4-ATF6 fusion system, where the DNA-binding domain of GAL4 is fused to ATF6, and activity is measured using a GAL4-responsive luciferase reporter [34]. Combining these reporter assays with protein analysis techniques offers a comprehensive assessment of ATF6 signaling from activation to functional outcomes.
Research Applications and Pathophysiological Context
The experimental methodologies for analyzing ATF6 activation have revealed its significance in various physiological and pathophysiological contexts. In neural development and function, ATF6-mediated UPR signaling contributes to synaptic plasticity, and its dysregulation has been implicated in neuropsychiatric disorders including autism spectrum disorders, schizophrenia, and bipolar disorder [32]. In sensory systems, loss-of-function mutations in ATF6 cause cone dysfunction disorders such as achromatopsia, and recent evidence indicates that ATF6 deficiency also leads to progressive sensorineural hearing loss, representing a blindness-deafness syndrome targeting hair cells and cone photoreceptors [37] [38].
In cancer biology, ATF6 activation promotes tumor cell survival under microenvironmental stress and contributes to therapy resistance, making it an attractive therapeutic target [17]. Interestingly, in colorectal cancer models, enforced activation of either ATF6 or XBP1 reduced cancer cell proliferation and stemness by activating PERK-eIF2α signaling, revealing unexpected cross-talk between UPR branches [36]. These diverse pathophysiological roles highlight the importance of precise ATF6 regulation and suggest potential therapeutic applications for modulators of ATF6 signaling across multiple disease contexts.
The strategic inhibition of ATF6 is being explored in oncology, with compounds like Ceapins specifically blocking ATF6 activation by inhibiting its traffic to the Golgi apparatus [17]. Conversely, small molecule activators of ATF6 could potentially ameliorate pathologies associated with protein misfolding, such as neurodegenerative diseases. As research continues to elucidate the nuanced functions of ATF6 in different tissues and disease states, the experimental approaches outlined in this review will remain fundamental to advancing both basic science and translational applications.
The endoplasmic reticulum (ER) serves as a crucial protein-folding compartment and dynamic calcium store, making it exceptionally sensitive to disturbances in intracellular homeostasis [39]. Various biochemical, physiological, and pathological stimuli—including calcium imbalance, redox status alterations, glucose deprivation, and increased protein synthesis demands—can disrupt ER function, leading to the accumulation of unfolded or misfolded proteins [39] [40]. To counteract this proteostatic imbalance, cells activate the unfolded protein response, an integrated signaling network orchestrated by three ER transmembrane sensors: PERK, IRE1, and ATF6 [39]. Initially, the UPR implements adaptive measures to restore ER homeostasis by transiently attenuating protein translation, enhancing ER folding capacity, and promoting degradation of misfolded proteins [39] [3]. However, when ER stress persists unresolved, the UPR undergoes a critical transition from pro-survival to pro-apoptotic signaling. This molecular switch represents a pivotal commitment point in cellular fate, with profound implications for the pathophysiology of numerous diseases, including diabetes, neurodegeneration, and cancer [40] [41] [3].
The Initial Adaptive Phase of the UPR
The Tripartite Signaling Network
Under non-stress conditions, all three UPR sensors remain inactive through their association with the ER chaperone GRP78/BiP. The accumulation of unfolded proteins triggers BiP dissociation, leading to sequential activation of the UPR arms [39] [3]:
PERK-eIF2α Pathway: Activated PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α), leading to global translational attenuation that reduces the protein-folding load on the stressed ER. This translational control is paradoxically coupled with selective translation of specific mRNAs, including the transcription factor ATF4, which activates genes involved in antioxidant response and amino acid metabolism [39] [40].
IRE1-XBP1 Pathway: IRE1, possessing both kinase and endoribonuclease activities, undergoes oligomerization and autophosphorylation upon activation. Its RNase domain mediates the unconventional splicing of XBP1 mRNA, generating a potent transcription factor that upregulates genes encoding ER chaperones, folding enzymes, and components of ER-associated degradation [39] [3].
ATF6 Pathway: Following BiP dissociation, ATF6 translocates to the Golgi apparatus where it undergoes proteolytic cleavage by site-1 and site-2 proteases. The released cytosolic fragment functions as a transcription factor that migrates to the nucleus and enhances expression of ER quality control components, often in coordination with XBP1 [39] [42].
Table 1: The Three Arms of the Unfolded Protein Response
| UPR Arm | Sensor Activation | Key Effectors | Primary Adaptive Functions |
|---|---|---|---|
| PERK | Homodimerization and autophosphorylation after BiP dissociation | p-eIF2α, ATF4, NRF2 | Translational attenuation; Antioxidant response; Expression of chaperones |
| IRE1 | Oligomerization and trans-autophosphorylation | XBP1s, TRAF2, JNK | Transcriptional upregulation of folding and degradation machinery; ER biogenesis |
| ATF6 | Golgi translocation and proteolytic cleavage | ATF6f (p50) | Enhancement of ER quality control and chaperone expression |
Integrated Stress Resolution Mechanisms
The adaptive UPR coordinates multiple mechanisms to resolve protein-folding defects. ER-associated degradation identifies misfolded proteins, retrotranslocates them to the cytoplasm, and targets them for proteasomal degradation [3]. Additionally, ER stress activates autophagy pathways, including chaperone-mediated autophagy and reticulophagy, which clear protein aggregates and damaged organelles [43] [44]. The transcription factors XBP1s, ATF6f, and ATF4 collaboratively regulate genes encoding chaperones, foldases, and lipid biosynthetic enzymes to expand the ER's functional capacity [39] [42]. This integrated response provides a temporary survival advantage, allowing cells to adapt to transient stress conditions.
The Molecular Switch to Apoptosis
When ER stress is prolonged or severe, cellular protective mechanisms are overwhelmed, leading to a strategic transition toward apoptosis. This molecular switch involves multiple interconnected pathways that converge on the mitochondrial apoptosis machinery.
Transcriptional Control via CHOP
The transcription factor C/EBP homologous protein plays a central role in ER stress-induced apoptosis. CHOP expression is tightly regulated by the PERK-eIF2α-ATF4 pathway and, to a lesser extent, by other UPR arms [39] [40]. Under persistent ER stress, CHOP orchestrates a pro-apoptotic program through several mechanisms:
- Downregulation of Bcl-2: CHOP suppresses expression of the anti-apoptotic protein Bcl-2, thereby promoting mitochondrial outer membrane permeabilization [39].
- ER Oxidase Activation: CHOP transcriptionally activates ERO1α, an ER oxidase that promotes hyperoxidization of the ER environment, ultimately leading to calcium release and mitochondrial dysfunction [39].
- Protein Synthesis Regulation: CHOP induces growth arrest and DNA damage-inducible protein 34, which promotes dephosphorylation of eIF2α and restoration of protein synthesis, paradoxically increasing the protein-folding load on the already stressed ER [3].
IRE1-Dependent Apoptotic Signaling
Under irremediable ER stress, IRE1 signaling shifts from adaptive to pro-apoptotic through two principal mechanisms:
- TRAF2-ASK1-JNK Pathway: Activated IRE1 recruits TNF receptor-associated factor 2, which serves as a platform for apoptosis signal-regulating kinase 1. ASK1 subsequently activates JNK, which phosphorylates and inhibits Bcl-2 while enhancing the pro-apoptotic activity of BH3-only proteins like BIM [39] [40].
- Regulated IRE1-Dependent Decay: Under severe ER stress, IRE1's RNase activity becomes less specific and can degrade ER-localized mRNAs, potentially contributing to apoptosis induction [45].
ER-Mitochondrial Cross-Talk
Calcium-mediated signaling forms a critical connection between ER stress and mitochondrial apoptosis. Pro-apoptotic Bcl-2 family members, Bax and Bak, permeabilize the ER membrane, leading to calcium release into the cytosol [39]. This calcium is taken up by mitochondria, causing depolarization of the mitochondrial membrane, permeability transition, and cytochrome c release. The calcium-dependent protease calpain activates caspase-12 (in rodents), which then activates the caspase cascade [39]. In humans, where caspase-12 is largely nonfunctional, ER stress-induced caspase activation occurs primarily through mitochondrial cytochrome c release and apoptosome formation [39].
Table 2: Key Pro-apoptotic Mechanisms in ER Stress
| Mechanism | Key Mediators | Downstream Effects |
|---|---|---|
| CHOP-mediated Transcription | ERO1α, GADD34, DR5, TRB3 | ER hyperoxidation; Protein synthesis overload; Death receptor signaling |
| IRE1-TRAF2 Signaling | ASK1, JNK | Bcl-2 inhibition; BIM activation; Mitochondrial apoptosis |
| Calcium-mediated Apoptosis | Bax/Bak, Calpain, Caspase-12 | Mitochondrial membrane permeabilization; Caspase activation |
| BH3-only Protein Induction | NOXA, BIM, PUMA | BAX/BAK activation; Mitochondrial outer membrane permeabilization |
Quantitative Dynamics of the UPR Switch
The transition from adaptation to apoptosis is governed by kinetic and quantitative aspects of UPR signaling. The duration and intensity of ER stress determine the balance between pro-survival and pro-death outputs.
Table 3: Temporal Dynamics of UPR Signaling Components
| Signaling Component | Early Adaptive Response (0-8h) | Late Apoptotic Response (>12h) |
|---|---|---|
| PERK signaling | Transient eIF2α phosphorylation; ATF4 translation | Sustained eIF2α phosphorylation; CHOP induction |
| IRE1 activity | Specific XBP1 splicing; Chaperone induction | RIDD activation; TRAF2-ASK1-JNK pathway engagement |
| ATF6 signaling | Proteolytic activation; ERSE-driven transcription | Declining activity; Secondary to PERK and IRE1 |
| Cellular Outcome | Adaptation and survival | Commitment to apoptosis |
Experimental Approaches for Monitoring the UPR Switch
Research Reagent Solutions
Table 4: Essential Reagents for UPR Research
| Reagent / Method | Function / Target | Application in UPR Research |
|---|---|---|
| Thapsigargin | SERCA pump inhibitor | ER stress inducer; Causes calcium disruption |
| Tunicamycin | N-linked glycosylation inhibitor | ER stress inducer; Impairs protein folding |
| sUPRa Reporter | BiP promoter activity | Global UPR monitoring with cellular resolution |
| XBP1 Splicing Assay | IRE1 RNase activity | Detection of IRE1 activation; Adaptive UPR assessment |
| CHOP Reporter Systems | CHOP promoter activity | Apoptotic UPR signaling measurement |
| JNK Inhibitors | JNK activity | Dissection of IRE1-TRAF2-ASK1-JNK pathway |
| 4μ8c | IRE1 RNase inhibitor | Selective inhibition of IRE1 signaling branch |
Methodologies for Detection of ER Stress and Apoptosis
Cell Culture and Stress Induction: Mammalian cells are maintained in appropriate media supplemented with fetal bovine serum. For ER stress induction, cells are treated with thapsigargin (500 μM stock in DMSO, working concentration typically 0.5-2 μM) or tunicamycin (10 mg/mL stock in DMSO, working concentration 2-10 μg/mL) for varying durations [39].
Western Immunoblot Analysis: Cells are lysed using modified RIPA buffer supplemented with protease and phosphatase inhibitors. Key targets for immunoblotting include phospho-eIF2α, ATF4, CHOP, XBP1s, phospho-JNK, and cleaved caspases. PERK and IRE1 activation can be assessed through immunoprecipitation of phosphorylated forms [39].
RNA Analysis: XBP1 splicing is monitored by RT-PCR using specific primers that distinguish unspliced (XBP1u) from spliced (XBP1s) variants based on size difference. Quantitative PCR can measure transcript levels of UPR target genes such as BiP, CHOP, and ERdj4 [39] [46].
Global UPR Monitoring with sUPRa: The novel sUPRa reporter system utilizes a 195-bp fragment of the mouse BiP promoter driving expression of mNeonGreen, with normalization to constitutively expressed mScarlet. This dual-color system provides sensitive detection of global UPR activity with single-cell resolution, outperforming single-pathway reporters [46].
UPR Signaling Pathways Visualization
UPR Fate Decision Network: This diagram illustrates the parallel signaling pathways that mediate the transition from adaptive to apoptotic UPR. Under initial ER stress, all three sensors promote adaptive responses. With prolonged or severe stress, the balance shifts toward pro-apoptotic signaling through CHOP induction, IRE1-TRAF2-ASK1-JNK activation, and calcium-mediated apoptosis.
Therapeutic Implications and Future Directions
Understanding the molecular switch of the UPR has significant therapeutic implications. In cancer, therapeutic resistance can emerge from the ability of tumor cells to leverage the adaptive UPR to survive microenvironmental stress [45]. Similarly, in diabetes, the delicate balance between adaptive and apoptotic UPR in pancreatic β-cells influences disease progression [41]. Emerging therapeutic strategies aim to modulate this switch by targeting specific UPR components—either to enhance apoptosis in cancer cells or to promote survival in degenerative diseases [3] [45]. The development of highly sensitive UPR reporters, such as sUPRa, provides new opportunities to quantify physiological UPR activation and screen for compounds that can precisely manipulate this critical cellular fate decision [46]. As our understanding of the UPR switch deepens, so does our potential to develop targeted therapies for the numerous diseases characterized by ER proteostasis imbalance.
The endoplasmic reticulum (ER) serves as a critical cellular organelle for protein folding, lipid synthesis, and calcium homeostasis. Its proper functioning is essential for cellular viability, and disruptions to its activity trigger a state known as ER stress. This technical guide examines the principal physiological inducers of ER stress—hypoxia, nutrient deprivation, and metabolic demands—within the broader context of unfolded protein response (UPR) research. We detail the molecular mechanisms through which these stressors disrupt ER proteostasis, explore the resulting signaling cascades, and discuss the implications for disease pathogenesis and therapeutic development. The content is structured to provide researchers, scientists, and drug development professionals with a comprehensive resource, complete with experimental protocols, visualization of key pathways, and essential research reagents.
The endoplasmic reticulum is a multifaceted organelle responsible for the synthesis, folding, and modification of approximately one-third of the cellular proteome, alongside critical roles in lipid synthesis, steroid biosynthesis, carbohydrate metabolism, and calcium storage [1] [19]. The ER provides a specialized environment rich in chaperones and enzymes to ensure proper protein folding and assembly. When the protein-folding capacity of the ER is overwhelmed by intrinsic or extrinsic stressors, the condition termed "ER stress" ensues, characterized by the accumulation of unfolded or misfolded proteins within the ER lumen [3] [19].
To counteract ER stress and restore proteostasis, cells activate an integrated signal transduction pathway known as the unfolded protein response (UPR). The UPR is orchestrated by three principal ER-transmembrane sensors: protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) [47] [3] [48]. Under normal, non-stress conditions, these sensors are maintained in an inactive state through association with the ER chaperone BiP (GRP78). The accumulation of unfolded proteins triggers the dissociation of BiP, leading to the activation of each sensor [48].
- PERK Pathway: Activated PERK phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α), leading to a rapid but transient attenuation of global protein synthesis to reduce the incoming protein load on the ER. Paradoxically, this phosphorylation selectively promotes the translation of specific mRNAs, including that of activating transcription factor 4 (ATF4), which upregulates genes involved in amino acid metabolism, antioxidant response, and ER stress-induced apoptosis [3] [19].
- IRE1 Pathway: IRE1 possesses both kinase and endoribonuclease activities. Its activation leads to the unconventional splicing of XBP1 mRNA, generating a potent transcription factor (spliced XBP1, XBP1s) that drives the expression of genes encoding ER chaperones, lipid biosynthesis enzymes, and components of ER-associated degradation (ERAD) [1] [3].
- ATF6 Pathway: ER stress triggers the translocation of ATF6 to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P and S2P). The released cytosolic fragment (ATF6f) translocates to the nucleus and enhances the transcription of ER chaperones and XBP1 [3] [49].
The primary objective of the UPR is to restore ER homeostasis by reducing protein translation, enhancing the ER's protein-folding capacity, and clearing misfolded proteins. However, under severe or prolonged stress conditions, the UPR can initiate apoptotic signaling, eliminating severely damaged cells [3] [48]. The following sections will dissect how specific physiological stressors—hypoxia, nutrient deprivation, and metabolic demands—activate this complex response and detail the experimental approaches used to investigate them.
Hypoxia as a Key Inducer of ER Stress
Molecular Mechanisms Linking Hypoxia and ER Stress
Hypoxia, a condition of low oxygen tension (often defined as <2% O₂ in solid tumors), is a potent inducer of ER stress. The molecular mechanisms underlying this induction are multifaceted. Molecular oxygen is a essential cofactor for the formation of disulfide bonds, a critical step in the proper folding of many proteins within the ER. Hypoxia directly disrupts this process, leading to an accumulation of unfolded and misfolded proteins that activates the UPR [50] [48]. Furthermore, hypoxia stabilizes the transcription factor hypoxia-inducible factor-1α (HIF-1α), which orchestrates a broad transcriptional program supporting cellular adaptation to low oxygen. There is significant crosstalk between the hypoxic response and the UPR, creating an integrated adaptive network [50] [51] [48].
Table 1: Key Molecular Interactions Between Hypoxia and ER Stress Pathways
| Hypoxia-Induced Factor/Pathway | Interaction with UPR | Functional Outcome |
|---|---|---|
| Disulfide Bond Disruption | Accumulation of unfolded proteins; Activation of PERK, IRE1, ATF6 | UPR initiation to manage proteotoxicity [48] |
| HIF-1α Stabilization | UPR components (ATF4, XBP1) upregulate Siah2, degrading PHDs and further stabilizing HIF-1α | Positive feedback loop enhancing hypoxic adaptation [48] |
| ATF4 (UPR) | Binds to VEGF promoter; Potentiates HIF-1 transactivation | Synergistic upregulation of pro-angiogenic factors like VEGF and BNIP3 [51] |
| XBP1s (UPR) | Forms a transcriptional complex with HIF-1α | Co-regulation of genes critical for survival in hypoxic microenvironments [48] |
Experimental Protocols for Hypoxia Research
Protocol: Investigating ER Stress under Hypoxic Conditions in Cell Culture
- Hypoxia Chamber Setup: Place cultured cells (e.g., cancer cell lines, macrophages) in a specialized hypoxia chamber or incubator. Flush the chamber with a pre-mixed gas mixture containing 1% O₂, 5% CO₂, and balance N₂. Maintain humidity to prevent evaporation of culture media. Standard time points range from 2 to 24 hours [50] [48].
- Chemical Induction (Alternative): As a more accessible alternative, treat cells with chemical hypoxia mimetics such as Cobalt Chloride (CoCl₂) at a concentration of 100-200 µM for 24 hours. CoCl₂ stabilizes HIF-1α by inhibiting prolyl hydroxylases (PHDs) [48].
- Sample Collection: Harvest cells for downstream analysis. For protein extraction, use RIPA buffer supplemented with protease and phosphatase inhibitors. For RNA extraction, use TRIzol or commercial silica-membrane columns.
- Downstream Analysis:
- Western Blotting: Probe for HIF-1α, phosphorylated PERK (p-PERK), phosphorylated eIF2α (p-eIF2α), ATF4, CHOP, and XBP1s. BiP/GRP78 is a general marker of UPR activation.
- Quantitative PCR (qPCR): Measure mRNA levels of UPR target genes such as BiP, CHOP, XBP1s, and hypoxic targets like VEGFA and BNIP3 [51].
- Immunofluorescence: Visualize the nuclear translocation of HIF-1α, ATF4, or XBP1s using specific antibodies and confocal microscopy.
Figure 1: Molecular Crosstalk Between Hypoxia and ER Stress Pathways. Hypoxia disrupts disulfide bonding and stabilizes HIF-1α, leading to UPR activation. The UPR branches, through ATF4 and XBP1s, upregulate Siah2, which degrades PHDs, creating a positive feedback loop that further stabilizes HIF-1α. HIF-1α and ATF4 can then synergistically activate the transcription of target genes like VEGF and BNIP3.
Nutrient Deprivation and Metabolic Demands
Glucose Deprivation and Lipotoxicity
Nutrient fluctuations pose a significant challenge to ER homeostasis. The ER is a key nutrient-sensing organelle, and imbalances directly impact its function.
- Glucose Deprivation (Glucotoxicity): The ER is highly dependent on ATP and glucose for its chaperone functions and for the glycosylation of nascent proteins. Glucose deprivation leads to an energy crisis and impaired glycosylation, resulting in the accumulation of unmodified, unfolded proteins that trigger the UPR [52] [19]. This is particularly relevant in conditions like diabetes and ischemia.
- Lipotoxicity: Excess saturated fatty acids (SFAs), such as palmitate, are potent inducers of ER stress, especially in metabolic tissues like the liver, pancreas, and adipose tissue. SFAs can disrupt the physical properties of ER membranes, impair the function of ER calcium pumps (SERCA), and directly saturate the protein-folding machinery, leading to chronic UPR activation [52] [19] [53]. In macrophages, SFAs engage the IRE1α pathway to activate the NLRP3 inflammasome, driving the production of the pro-inflammatory cytokine IL-1β, a key link between ER stress and metabolic inflammation [19].
Calcium Homeostasis and Energetic Demands
Calcium (Ca²⁺) is a critical cofactor for many ER chaperones, including calnexin and calreticulin. The ER lumen maintains a high Ca²⁺ concentration, and its depletion is a classic inducer of ER stress. Agents like thapsigargin, a specific inhibitor of the SERCA pump, are routinely used in research to deplete ER Ca²⁺ stores and induce robust UPR activation [3] [19]. Furthermore, the high metabolic and secretory activity of specialized cells, such as pancreatic β-cells (insulin), hepatocytes (albumin, lipids), and plasma cells (antibodies), imposes a constant, substantial demand on the ER's folding capacity. Any increase in demand or perturbation in energy supply can easily tilt the balance toward ER stress [52] [53].
Table 2: Nutrient and Metabolic Inducers of ER Stress
| Stressor | Molecular Insult | Experimental Agent(s) | Key UPR Pathways Engaged |
|---|---|---|---|
| Glucose Deprivation | ATP depletion; Impaired N-linked glycosylation | Culture in low-glucose/no-glucose media; 2-Deoxy-D-glucose (2-DG) [52] | PERK-eIF2α-ATF4; IRE1-XBP1 |
| Lipotoxicity | SFA incorporation into membranes; Disruption of ER Ca²⁺ homeostasis; Saturation of folding capacity | Palmitate (250-500 µM conjugated to BSA) [19] | IRE1α (inflammasome activation); PERK-ATF4-CHOP |
| ER Ca²⁺ Depletion | Loss of Ca²⁺-dependent chaperone function | Thapsigargin (1-5 µM) [3] [19] | All three branches (PERK, IRE1, ATF6) strongly activated |
| Increased Secretory Demand | Overwhelming of folding and trafficking capacity | Genetic models; Cytokine stimulation in immune cells [52] [47] | IRE1-XBP1 (primary pathway for ER expansion) |
Experimental Protocols for Nutrient Stress
Protocol: Inducing and Assessing ER Stress via Nutrient Deprivation and Lipotoxicity
Glucose Deprivation:
- Grow cells to 70-80% confluency in standard high-glucose medium (e.g., 25 mM Glucose).
- Rinse cells with PBS and replace the medium with glucose-free medium supplemented with dialyzed fetal bovine serum (FBS) to remove confounding sugars. Include a control group with normal glucose.
- Incubate for 2-16 hours before harvesting for analysis [52].
Lipotoxicity Induction with Palmitate:
- Palmitate-BSA Conjugate Preparation: Dissolve sodium palmitate in sterile water by heating to 70°C. Conjugate it to fatty acid-free BSA in serum-free medium at a 6:1 or 8:1 molar ratio (Palmitate:BSA) by shaking at 37°C for 1 hour. Prepare a BSA-only control solution.
- Cell Treatment: Dilute the palmitate-BSA conjugate or BSA control in complete cell culture medium to a final palmitate concentration of 250-500 µM. Treat cells for 12-24 hours [19].
- Downstream Analysis: Assess UPR activation via Western blot (p-eIF2α, CHOP, XBP1s) and qPCR (BiP, CHOP). To investigate inflammation, measure IL-1β and IL-6 secretion by ELISA.
Research Toolkit: Reagents and Experimental Solutions
Table 3: Essential Research Reagents for ER Stress Investigation
| Reagent / Tool | Function / Mechanism of Action | Primary Research Application |
|---|---|---|
| Tunicamycin (Tm) | Inhibits N-linked glycosylation, preventing proper protein folding [19] | A classic and potent inducer of ER stress via impaired protein processing. |
| Thapsigargin (Tg) | Inhibits the SERCA pump, depleting ER calcium stores [3] [19] | A robust and reliable inducer of all three UPR branches via disruption of Ca²⁺ homeostasis. |
| 4-Phenylbutyric Acid (4-PBA) | Chemical chaperone that stabilizes protein conformation [53] | Used to alleviate ER stress and improve proteostasis in models of metabolic disease. |
| Tauroursodeoxycholic Acid (TUDCA) | Chemical chaperone that reduces protein misfolding [53] | Ameliorates ER stress in models of obesity, diabetes, and NAFLD. |
| STF-083010 | Selective inhibitor of IRE1α's RNase activity [19] | Used to dissect the specific role of the IRE1-XBP1/RIDD pathway in inflammation and disease. |
| ISRIB | Reverses the effects of eIF2α phosphorylation, restoring translation [3] | Used to inhibit the PERK-ATF4 pathway and study its functional consequences. |
| BSA-Conjugated Palmitate | Delivers saturated fatty acids to cells to model lipotoxicity [19] | Key reagent for studying ER stress in the context of obesity and metabolic syndrome. |
| Anti-BiP/GRP78 Antibody | Detects levels of the central ER chaperone and UPR master regulator. | Standard marker for UPR activation in Western blot, immunofluorescence, and immunoprecipitation. |
| Anti-CHOP Antibody | Detects the pro-apoptotic transcription factor downstream of PERK-ATF4. | Key marker for the transition from adaptive UPR to pro-apoptotic signaling. |
Figure 2: ER Stress Integration of Metabolic Signals. Various metabolic stressors, including nutrient imbalance and high secretory demand, converge on UPR activation. The outcome depends on the intensity and duration of stress. An adaptive UPR restores homeostasis, but chronic stress leads to apoptosis or metabolic inflammation, driving systemic metabolic dysfunction.
The in-depth understanding of ER stress inducers has profound therapeutic implications. Chronic, unresolved ER stress is a key pathological feature in a wide range of conditions, including type 2 diabetes, obesity, non-alcoholic fatty liver disease (NAFLD), neurodegenerative disorders, and cancer [52] [3] [53]. In metabolic diseases, ER stress in insulin-sensitive tissues and pancreatic β-cells contributes to insulin resistance and the failure of insulin secretion, respectively [53]. The link between ER stress and inflammation, particularly through mechanisms like IRE1α-driven NLRP3 inflammasome activation in macrophages, establishes a vicious cycle that perpetuates metabolic syndrome [19] [48].
Consequently, the UPR pathways present attractive therapeutic targets. Strategies include:
- Chemical Chaperones: Drugs like TUDCA and 4-PBA have shown efficacy in preclinical models to alleviate ER stress and improve glucose homeostasis and insulin sensitivity [53].
- Targeted UPR Modulators: Developing specific inhibitors for individual UPR branches, such as IRE1α RNase inhibitors, holds promise for curbing maladaptive UPR in specific disease contexts without completely abrogating this vital survival pathway [19].
- Lifestyle and Nutritional Interventions: Given that overnutrition and saturated fats are key inducers, dietary modifications remain a foundational approach to reducing systemic ER stress.
In conclusion, hypoxia, nutrient deprivation, and metabolic demands represent core physiological inducers of ER stress. They disrupt the delicate proteostatic balance within the ER, activating the complex signaling network of the UPR. The decision between adaptive survival and pathological apoptosis depends on the intensity and duration of the stress signal. Continued research into the precise mechanisms of these inducers and their crosstalk is paramount for developing novel therapeutics aimed at modulating the UPR to treat a vast spectrum of human diseases.
Detecting and Modulating UPR: From Laboratory Techniques to Therapeutic Exploitation
The unfolded protein response (UPR) is an essential adaptive signaling network activated by the accumulation of misfolded proteins within the endoplasmic reticulum (ER), a condition known as ER stress. In disease states, including cancer and metabolic disorders, the UPR can be aberrantly activated, driving pathogenesis and influencing treatment responses [54] [55] [56]. Accurately measuring UPR activation is therefore critical for both basic research and drug development. Given the complexity and dynamic nature of this pathway, a multi-marker approach that simultaneously probes multiple UPR branches at the RNA and protein level provides the most comprehensive and reliable assessment. This technical guide outlines best practices for such an integrated analysis, providing researchers with detailed methodologies to capture the full spectrum of UPR activity.
The Core UPR Signaling Network
In mammalian cells, the UPR is initiated by three ER-transmembrane sensors: IRE1α, PERK, and ATF6. Under homeostatic conditions, these sensors are largely bound to the ER chaperone BiP (GRP78). An accumulation of unfolded proteins leads to BiP dissociation and subsequent activation of each sensor, triggering a multifaceted transcriptional and translational reprogramming aimed at restoring proteostatic balance [57] [58].
- IRE1α Branch: Activated IRE1α exhibits endoribonuclease activity, catalyzing the unconventional splicing of XBP1 mRNA. This splicing event removes a 26-nucleotide intron, causing a frameshift that produces a potent transcription factor, spliced XBP1 (XBP1s), which upregulates genes involved in ER biogenesis and ER-associated degradation (ERAD) [57] [58].
- PERK Branch: Activated PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α). This global translational attenuation reduces the protein-folding load on the ER while paradoxically promoting the translation of specific mRNAs, such as that encoding the transcription factor ATF4. ATF4 drives the expression of genes related to amino acid metabolism, antioxidant response, and the pro-apoptotic factor CHOP [57] [56].
- ATF6 Branch: Following ER stress, full-length ATF6 (p90ATF6) translocates to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P, S2P). This releases its cytosolic fragment (p50ATF6), a transcription factor that activates ER chaperone genes and, cooperatively with XBP1s, components of the ERAD pathway [58].
The following diagram illustrates the core UPR signaling network and the key markers for its detection:
RNA-Based Analysis of UPR Activation
Monitoring changes at the transcript level can reveal early and specific activation of different UPR arms. The table below summarizes the key RNA markers and their significance.
Table 1: Key RNA Markers for UPR Analysis
| Target | Detection Method | Significance & Notes |
|---|---|---|
| XBP1 Splicing | RT-PCR, RNA-seq | Gold standard for IRE1α activation. Primer sets distinguish unspliced (XBP1u) and spliced (XBP1s) isoforms. A shift to the spliced band indicates activation [58] [56]. |
| ATF4 mRNA | qPCR, RNA-seq | Upregulated transcriptionally, but protein synthesis is primarily controlled by translational mechanisms. Useful but not definitive alone [56]. |
| CHOP (DDIT3) mRNA | qPCR, RNA-seq | A robust PERK-ATF4 pathway target. Strong transcriptional induction makes it a sensitive PERK branch marker [58]. |
| BiP (HSPA5) mRNA | qPCR, RNA-seq | A general ER stress marker. Induced by multiple UPR branches; indicates adaptive UPR and chaperone upregulation [57] [58]. |
| ERAD Component mRNAs | qPCR, RNA-seq | Targets like EDEM1 and HERP are induced by XBP1s/ATF6. Indicate a later, adaptive UPR phase focused on clearance [57]. |
Detailed Experimental Protocol: XBP1 Splicing Analysis by RT-PCR
This protocol is a cornerstone for assessing IRE1α activity [58].
- RNA Extraction & DNase Treatment: Isolate high-quality total RNA using a guanidinium thiocyanate-phenol-chloroform-based method. Treat samples with DNase I to remove genomic DNA contamination.
- cDNA Synthesis: Reverse transcribe 0.5–1 µg of total RNA using random hexamers or oligo(dT) primers and a reverse transcriptase enzyme.
- PCR Amplification:
- Use primers flanking the 26-nucleotide intron in human XBP1:
- Forward:
5'-TTACGAGAGAAAACTCATGGCC-3' - Reverse:
5'-GGGTCCAAGTTGTCCAGAATGC-3'
- Forward:
- Perform PCR with a standard Taq polymerase for 30-35 cycles.
- The expected PCR products are:
- Unspliced XBP1 (XBP1u): 289 bp
- Spliced XBP1 (XBP1s): 263 bp
- Use primers flanking the 26-nucleotide intron in human XBP1:
- Product Visualization: Resolve the PCR products on a 2.5–3% agarose gel. Activation of IRE1α is indicated by a clear band shift from the unspliced to the spliced form, or the presence of both.
Protein-Based Analysis of UPR Activation
Protein-level analysis confirms pathway activation and reveals functional outcomes, including translational control and post-translational modifications. The table below summarizes the key protein markers.
Table 2: Key Protein Markers for UPR Analysis
| Target | Detection Method | Significance & Notes |
|---|---|---|
| Phospho-eIF2α (Ser51) | Western Blot | Direct marker of PERK kinase activity. Use phospho-specific antibodies. Compare to total eIF2α for loading [58] [56]. |
| ATF4 | Western Blot | Accumulates rapidly post-stress due to translational upregulation. A key PERK branch output [56]. |
| CHOP | Western Blot, IHC | Strongly induced at the protein level following transcriptional upregulation. A marker of prolonged/severe ER stress [58]. |
| Cleaved ATF6 (p50) | Western Blot | Technically challenging due to low abundance and lack of high-quality antibodies. Indirect monitoring via target genes is often preferred [58]. |
| BiP/GRP78 | Western Blot | A general ER stress marker. Increased expression indicates UPR-mediated chaperone induction [57] [56]. |
| XBP1s | Western Blot | Requires antibodies specific to the C-terminal domain unique to the spliced isoform. Confirms IRE1α activation and functional protein production [56]. |
Detailed Experimental Protocol: Western Blot Analysis for PERK Branch
This protocol outlines the steps to detect key proteins in the PERK-eIF2α-ATF4 axis [59] [58].
- Protein Lysate Preparation: Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. For phospho-protein detection, phosphatase inhibitors are critical.
- Protein Quantification & Gel Electrophoresis: Determine protein concentration using a BCA or Bradford assay. Load 20-40 µg of total protein per lane on a 4-12% Bis-Tris polyacrylamide gel and separate by SDS-PAGE.
- Protein Transfer & Blocking: Transfer proteins to a PVDF or nitrocellulose membrane. Block the membrane with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature.
- Antibody Incubation:
- Incubate with primary antibodies diluted in blocking buffer overnight at 4°C.
- Recommended primary antibodies:
- Anti-phospho-eIF2α (Ser51)
- Anti-total eIF2α (loading control)
- Anti-ATF4
- Anti-CHOP
- Wash membrane 3x for 5-10 minutes with TBST.
- Incubate with appropriate HRP-conjugated secondary antibodies for 1 hour at room temperature.
- Detection: Develop the blot using enhanced chemiluminescence (ECL) substrate and image with a digital imager. Expected results under ER stress include increased phospho-eIF2α, ATF4, and CHOP protein levels.
An Integrated Multi-Omics Workflow
For a systems-level understanding, a multi-omics approach that combines transcriptomic, proteomic, and translatomic (ribosome profiling) data is powerful. This can reveal the complex, temporal layers of UPR regulation, as demonstrated in a study on astrocytoma cells [56].
- Staged Sampling: The UPR is highly dynamic. Collect samples at multiple early and late time points (e.g., 2, 6, 16, and 24 hours) after ER stress induction to capture immediate translational responses and later transcriptional adaptations.
- Multi-Assay Parallelism:
- Ribosome Profiling: Reveals global translational attenuation and identifies specific mRNAs (like ATF4) undergoing preferential translation during stress.
- RNA-seq: Quantifies genome-wide changes in transcript abundance, identifying UPR target genes and splicing events (like XBP1).
- Shotgun Proteomics: Confirms changes in the abundance of key UPR-related proteins and metabolic enzymes, validating functional outcomes.
- Data Integration: Integrated analysis can identify the "UPR regulon"—a core set of genes consistently induced across omics layers—and uncover novel, clinically relevant metabolic adaptations, such as UPR-induced resistance to antifolate drugs [56].
The workflow for such an integrated study can be visualized as follows:
The Scientist's Toolkit: Essential Research Reagents
Successful UPR analysis depends on high-quality, validated reagents. The table below lists essential materials for key experiments.
Table 3: Essential Research Reagents for UPR Analysis
| Reagent / Assay | Function / Target | Specific Example / Note |
|---|---|---|
| ER Stress Inducers | Chemically induce ER stress for experimental studies. | Tunicamycin (N-glycosylation inhibitor), Thapsigargin (SERCA pump inhibitor), Brefeldin A (protein transport disruptor) [56]. |
| Phospho-Specific Antibodies | Detect activation-specific protein phosphorylation. | Anti-phospho-eIF2α (Ser51) for PERK branch; Anti-phospho-IRE1α (Ser724) (challenging to detect) [58]. |
| Transcription Factor Antibodies | Detect UPR-induced transcription factors and chaperones. | Anti-ATF4, Anti-CHOP, Anti-XBP1s (spliced isoform-specific), Anti-BiP/GRP78 [58] [56]. |
| qPCR/XBP1 Splicing Assays | Quantify mRNA levels and detect XBP1 splicing. | Validated primer sets for XBP1 (splicing), ATF4, CHOP, BiP, and HERP. Commercial XBP1 splicing assay kits are available [58]. |
| SomaScan Assay | Large-scale, high-throughput proteomic discovery. | Aptamer-based platform measuring ~7,000 proteins. Useful for unbiased biomarker discovery in biofluiments [60] [61]. |
| CIBERSORT Algorithm | Computational analysis of immune cell infiltration. | Used to correlate UPR signature gene expression with immune context, relevant for cancer and therapeutic studies [62]. |
The unfolded protein response (UPR) is an essential cellular signaling network that maintains proteostasis within the endoplasmic reticulum (ER). Upon ER stress, three sensor proteins—PERK, IRE1, and ATF6—orchestrate a multifaceted response ranging from adaptive mechanisms to apoptosis. For researchers investigating ER stress in contexts ranging from cancer to metabolic diseases, accurately measuring the activation of these pathways is paramount. This technical guide details the core readouts—phospho-PERK, XBP1 splicing, cleaved ATF6, and CHOP induction—that serve as definitive markers for UPR arm activation. These indicators provide a window into the cell's stress status, informing on strategic decisions in both basic research and drug development. The following sections will delineate the molecular basis, standard detection methodologies, and functional significance of each readout, supported by experimental data and practical protocols.
UPR Arm Readouts at a Glance
The table below summarizes the four key readouts, their molecular significance, and primary detection methods.
Table 1: Core UPR Readouts and Detection Strategies
| UPR Arm & Readout | Molecular Significance | Primary Detection Methods |
|---|---|---|
| PERK Arm: Phospho-PERK | Initial sensor autophosphorylation, rapidly indicates ER stress engagement. [63] [64] | Western blot (Phospho-specific antibodies), ELISA |
| IRE1 Arm: XBP1 Splicing | IRE1's endoribonuclease activity; produces potent transcription factor sXBP1. [65] [66] | RT-PCR (size difference), Western blot (sXBP1 antibody) |
| ATF6 Arm: Cleaved ATF6 | S1P/S2P protease-mediated activation; generates transcriptionally active p50ATF6 fragment. [66] [67] | Western blot (Anti-ATF6 N-terminal), Immunofluorescence |
| Integrated Output: CHOP Induction | Marker of severe/prolonged stress; transcriptionally regulated by all three arms. [64] [68] | Western blot (CHOP antibody), qRT-PCR, Immunofluorescence |
Detailed Analysis of UPR Readouts and Experimental Guidance
The PERK Arm and Phospho-PERK
The PERK pathway is one of the most rapidly activated arms of the UPR. Its activation is best measured by detecting phospho-PERK (Thr980), which reflects PERK autophosphorylation and dimerization upon ER stress. This event triggers the downstream PERK-eIF2α-ATF4 signaling cascade, ultimately leading to the induction of the pro-apoptotic transcription factor CHOP under persistent stress conditions [63] [68].
The functional significance of this pathway is context-dependent. In diabetic cardiomyopathy, the PERK pathway has been shown to play a more critical role in ROS-mediated ER stress-induced apoptosis compared to the IRE1 or ATF6 pathways [63]. Furthermore, the subcellular localization of PERK at mitochondria-associated ER membranes (MAMs) positions it as a key regulator in stress-induced communication between these two organelles [63].
Table 2: Experimental Guide for Phospho-PERK Detection
| Aspect | Protocol Details |
|---|---|
| Key Reagent | Anti-phospho-PERK (Thr980) antibody (e.g., Rabbit monoclonal) |
| Sample Prep | Harvest cells in RIPA buffer with phosphatase and protease inhibitors. |
| Protocol | 1. Resolve 20-50 µg protein via SDS-PAGE (8% gel).2. Transfer to PVDF membrane.3. Block with 5% BSA for 1 hour.4. Incubate with primary anti-phospho-PERK antibody (1:1000) overnight at 4°C.5. Incubate with HRP-conjugated secondary antibody (1:2000) for 1 hour.6. Detect with ECL reagent. Normalize to total PERK. |
| Controls | Positive Control: Cells treated with 2µM Thapsigargin for 2-4 hours.Negative Control: Untreated or PERK-knockdown cells. |
| Data Interpretation | A band at ~125 kDa indicates PERK phosphorylation. Quantify the ratio of p-PERK to total PERK. |
The IRE1 Arm and XBP1 Splicing
The IRE1 arm's most conserved and definitive output is the unconventional splicing of XBP1 mRNA. IRE1α's endoribonuclease activity removes a 26-nucleotide intron from XBP1u mRNA, leading to a frameshift and the production of a stable, potent transcription factor, spliced XBP1 (sXBP1) [65] [66]. This pathway is critical for adapting to ER stress, and sXBP1 drives the expression of genes involved in ER biogenesis and ER-associated degradation (ERAD).
The functional importance of XBP1 splicing extends to specialized secretory cells. While it is essential for plasma cell differentiation, its role is decoupled from other UPR elements like PERK [69]. Furthermore, in pathological contexts such as type 2 diabetes, ER stress-induced XBP1 splicing can transcriptionally upregulate human islet amyloid polypeptide (hIAPP), contributing to β-cell cytotoxicity [70].
Table 3: Experimental Guide for Detecting XBP1 Splicing
| Aspect | Protocol Details |
|---|---|
| Key Reagent | Specific primers flanking the splicing site: F: 5'-CCTTGTAGTTGAGAACCAGG-3', R: 5'-GGTCCAAGTTGTCCAGAATGC-3' [70]. |
| Sample Prep | Extract total RNA with TRI reagent; synthesize cDNA from 1µg RNA. |
| Protocol | 1. Perform standard PCR with the specific primers.2. Resolve PCR products on a 2.5-3% agarose gel.Expected Bands: XBP1u (unspliced): ~bp; XBP1s (spliced): ~bp (26bp smaller). |
| Controls | Positive Control: Cells treated with 10µg/mL Tunicamycin for 6-8 hours.Inhibitor Control: Use IRE1 RNase inhibitor (e.g., MKC3946, 10µM) to block splicing [70]. |
| Data Interpretation | The presence of the smaller band (XBP1s) indicates IRE1 activation. The ratio of XBP1s to XBP1u can be quantified. |
The ATF6 Arm and Cleaved ATF6
Activation of the ATF6 arm is characterized by the regulated intramembrane proteolysis of the full-length 90 kDa ATF6 (p90ATF6) to its cleaved, transcriptionally active 50 kDa fragment (p50ATF6). Under unstressed conditions, ATF6 is retained in the ER. Upon stress, it translocates to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P and S2P). The p50ATF6 fragment then moves to the nucleus to activate genes like XBP1 and CHOP [66] [64] [67].
It is important to note that some viruses have evolved mechanisms to subvert this canonical pathway. For instance, Enterovirus A71 infection can lead to atypical cleavage of ATF6, preventing the generation of the functional p50 fragment and hijacking the protein to support viral replication [67].
Table 4: Experimental Guide for Detecting Cleaved ATF6
| Aspect | Protocol Details |
|---|---|
| Key Reagent | Antibodies against the N-terminal transcription factor domain of ATF6 (e.g., Rabbit polyclonal anti-ATF6 aa 6-307) [67]. |
| Sample Prep | Prepare whole cell lysates in RIPA buffer. For nuclear fractionation, use a commercial kit to isolate nuclear proteins. |
| Protocol | 1. Resolve proteins on an 8-10% SDS-PAGE gel.2. Perform Western blot as standard.Expected Bands: p90ATF6 (full-length): ~90 kDa; p50ATF6 (cleaved/active): ~50 kDa. |
| Controls | Positive Control: Cells treated with 1µM Thapsigargin or 10µg/mL Tunicamycin for 4-8 hours.Localization Control: Compare whole lysate vs. nuclear fraction (p50 should enrich in nucleus). |
| Data Interpretation | The appearance of the p50 band indicates ATF6 activation. Loss of p90 may also be observed. |
The Apoptotic Switch and CHOP Induction
CHOP (C/EBP Homologous Protein, also known as GADD153) serves as a critical integrator of pro-apoptotic signals during severe or prolonged ER stress. While it is expressed at low levels under normal conditions, its transcription is robustly upregulated by all three UPR arms: via ATF4 downstream of PERK, by the cleaved ATF6 fragment, and indirectly via IRE1-XBP1 and IRE1-ASK1-p38MAPK/JNK pathways [64] [68]. CHOP promotes apoptosis by modulating the expression of BCL2 family proteins and regulating cellular redox status.
The role of CHOP can be highly tissue-specific. For example, in differentiating B lymphocytes (plasma cells), CHOP induction appears to be dispensable for apoptosis, and its deletion does not significantly alter cell lifespan or sensitivity to proteasome inhibitors [69]. This highlights the necessity of contextual interpretation when using CHOP as a marker for cell fate decisions.
Table 5: Experimental Guide for Monitoring CHOP Induction
| Aspect | Protocol Details |
|---|---|
| Key Reagent | Anti-CHOP antibody (e.g., Mouse monoclonal, Cell Signaling) [70]. |
| Sample Prep | Lyse cells in RIPA-DOC buffer with protease inhibitors. |
| Protocol | 1. Resolve 20-30 µg protein on a 10-12% SDS-PAGE gel.2. Transfer and block membrane.3. Incubate with anti-CHOP primary antibody (1:1000) overnight at 4°C.4. Proceed with standard ECL detection. Normalize to loading control (e.g., β-actin). |
| Controls | Positive Control: Cells treated with 2µM Thapsigargin or 10µg/mL Tunicamycin for 8-16 hours.Genetic Control: CHOP-knockout cells (if available) to confirm antibody specificity. |
| Data Interpretation | A band at ~29 kDa indicates CHOP protein. Its presence, especially after prolonged stress, signals a shift toward apoptosis. |
The Scientist's Toolkit: Essential Research Reagents
The table below compiles key reagents utilized in the studies cited within this guide, providing a practical resource for experimental design.
Table 6: Key Research Reagent Solutions for UPR Studies
| Reagent / Tool | Function / Target | Example Use Case |
|---|---|---|
| Tunicamycin | Inhibits N-linked glycosylation; induces ER stress. | General UPR inducer (e.g., 10 µg/mL for 6-24h) [71] [70]. |
| Thapsigargin | Sarco/ER Ca²⁺-ATPase (SERCA) inhibitor; induces ER stress by Ca²⁺ depletion. | General UPR inducer (e.g., 1-2 µM for 2-8h) [70]. |
| MKC3946 | Inhibits IRE1's RNase activity. | Blocks XBP1 splicing (e.g., 10 µM) [70]. |
| N-Acetylcysteine (NAC) | ROS scavenger. | Dissects ROS-dependent ER stress (e.g., 300 mg/kg in vivo) [63]. |
| siRNA (PERK, ATF6, IRE1) | Gene-specific knockdown. | Determines the specific role of each UPR sensor [63] [67]. |
| Phospho-specific Antibodies | Detect active, phosphorylated proteins. | Detecting phospho-PERK, phospho-eIF2α [63]. |
| Anti-CHOP Antibody | Detects CHOP protein induction. | Apoptosis marker in Western blot/IF [70]. |
UPR Signaling Pathway Integration and Crosstalk
The following diagram illustrates the core UPR pathways, highlighting the key readouts and their interrelationships, from initial stress sensing to functional outcomes like adaptation and apoptosis.
The precise measurement of phospho-PERK, XBP1 splicing, cleaved ATF6, and CHOP induction provides an unambiguous picture of UPR activation and the cellular decision point between survival and death. As research continues to reveal the context-dependent nuances of this pathway—from cancer cell adaptation to viral immune evasion—the standardized protocols and reagents outlined in this guide will serve as a critical foundation. For drug development professionals, these readouts offer valuable biomarkers for assessing therapeutic efficacy and mechanism of action, particularly for compounds designed to modulate ER stress in human disease. Future work will undoubtedly refine these tools further, enabling even more precise dissection and targeting of the UPR in pathophysiology.
The endoplasmic reticulum (ER) is a critical organelle for protein synthesis, folding, post-translational modification, and calcium storage in eukaryotic cells. Physiological or pathological disturbances that disrupt protein folding in the ER lumen lead to a state known as ER stress. To counteract this, cells activate a complex intracellular signaling network termed the unfolded protein response (UPR) [58] [3]. The primary objectives of the UPR are to restore proteostasis by reducing the protein load on the ER, increasing the organelle's folding capacity, and promoting the degradation of irreparably misfolded proteins. The UPR is initiated by three ER-resident transmembrane sensors: IRE1 (inositol-requiring enzyme 1), PERK (PKR-like ER kinase), and ATF6 (activating transcription factor 6) [58] [19]. Under normal conditions, these sensors are kept inactive through association with the ER chaperone BiP (GRP78). The accumulation of unfolded proteins causes BiP to dissociate, leading to the activation of all three pathways [58] [3]. If ER stress is too severe or prolonged, the UPR transitions from a pro-survival to a pro-apoptotic signaling cascade, ultimately leading to programmed cell death [3] [19].
For researchers studying these processes, specific chemical tools are indispensable for inducing ER stress in a controlled and reproducible manner. Tunicamycin, thapsigargin, and dithiothreitol (DTT) are three of the most widely utilized compounds for this purpose, each with a distinct molecular mechanism. This guide provides an in-depth technical overview of their use in ER stress and UPR research.
Molecular Mechanisms and Signaling Pathways
The following diagram illustrates the core UPR pathways and the specific points of action for tunicamycin, thapsigargin, and DTT.
Tunicamycin: Inhibitor of N-Linked Glycosylation
Tunicamycin is a nucleoside antibiotic isolated from Streptomyces sp. that potently inhibits the first committed step of N-linked protein glycosylation in the ER [72] [73]. Its primary molecular target is the enzyme UDP-N-acetylglucosamine:dolichyl-phosphate N-acetylglucosaminephosphotransferase (GPT) [72]. By structurally mimicking the substrate UDP-N-acetylglucosamine, tunicamycin competitively inhibits GPT, which normally catalyzes the transfer of GlcNAc-1-phosphate from UDP-GlcNAc to the lipid carrier dolichyl phosphate [74] [72]. This inhibition prevents the formation of dolichyl-diphosphate-N-acetylglucosamine, thereby blocking the entire subsequent pathway of N-linked glycan synthesis. Consequently, proteins that rely on N-glycosylation for their proper folding and stability fail to mature, leading to a robust accumulation of unfolded proteins in the ER lumen and the activation of all three branches of the UPR [72] [73].
Thapsigargin: Disruptor of Calcium Homeostasis
Thapsigargin is a sesquiterpene lactone extracted from the plant Thapsia garganica and is a highly potent and specific non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump [75] [76]. By binding to SERCA, thapsigargin blocks its ability to pump cytosolic calcium back into the ER lumen. This leads to a rapid and irreversible depletion of ER calcium stores and a corresponding increase in cytosolic calcium concentration [76]. Since calcium is an essential cofactor for many ER chaperones, including calnexin and calreticulin, its depletion disrupts critical protein-folding processes [3]. The resulting accumulation of misfolded proteins triggers ER stress and activates the UPR. Notably, because thapsigargin acts directly on the calcium pump, it induces ER stress without directly impairing protein glycosylation or promoting disulfide bond reduction [76] [19].
Dithiothreitol (DTT): Reducer of Disulfide Bonds
Dithiothreitol (DTT) is a small-molecule reducing agent that acts through a different mechanism. Its primary function is to reduce disulfide bonds [77]. In the oxidizing environment of the ER, disulfide bond formation is a crucial step in the correct folding and stabilization of many secreted and membrane proteins. DTT, with its two thiol groups, disrupts this process by reducing these disulfide bonds, thereby preventing proper protein folding and leading to the accumulation of unfolded proteins [77]. It is important to note that DTT is a broad-spectrum reducing agent and its effects are not confined to the ER; however, the high rate of disulfide bond formation in the ER makes this organelle particularly sensitive to DTT treatment. The reduction of disulfide bonds can also impact the activity of ER-resident enzymes and the oligomerization of UPR sensors themselves.
Comparative Profiling of ER Stress Inducers
The table below summarizes the key characteristics and experimental use parameters for tunicamycin, thapsigargin, and DTT.
Table 1: Comparative Profile of Common Chemical Inducers of ER Stress
| Parameter | Tunicamycin | Thapsigargin | Dithiothreitol (DTT) |
|---|---|---|---|
| Primary Molecular Target | GPT (GlcNAc-1-P-transferase) [72] | SERCA pump [76] | Disulfide bonds [77] |
| Core Mechanism of Action | Inhibits N-linked glycosylation [73] | Depletes ER calcium stores [76] | Reduces disulfide bonds [77] |
| Mode of Inhibition | Competitive for UDP-GlcNAc [72] | Non-competitive, irreversible [76] | Chemical reduction |
| Key Downstream Effects | Accumulation of non-glycosylated, misfolded proteins [74] | Disruption of calcium-dependent chaperone function [3] | Accumulation of unfolded proteins due to lack of disulfide bonds |
| Typical Working Concentration (Mammalian Cells) | 1 - 10 µg/mL [73] | 0.1 - 1 µM [19] | 1 - 5 mM |
| Common Treatment Duration | 4 - 16 hours [73] | 1 - 8 hours [19] | 30 minutes - 2 hours |
| Solubility & Storage | Soluble in DMSO, store at +4°C [73] | Soluble in DMSO, store as recommended by supplier | Soluble in water, unstable at basic pH, store at -20°C [77] |
Experimental Protocols and Applications
Standardized In Vitro Induction of ER Stress
This protocol outlines a general procedure for inducing ER stress in adherent mammalian cell lines.
Materials:
- Complete cell culture medium
- Serum-free medium or PBS for washing
- Tunicamycin stock (e.g., 1 mg/mL in DMSO) [73]
- Thapsigargin stock (e.g., 1 mM in DMSO) [19]
- DTT stock (e.g., 1 M in sterile water) [77]
- Dimethyl sulfoxide (DMSO), sterile
Method:
- Cell Seeding: Seed cells in appropriate culture vessels and allow them to adhere and grow to 60-80% confluence.
- Preparation of Working Solutions:
- Tunicamycin: Dilute the stock solution in complete medium to a final concentration typically between 1 and 10 µg/mL. A DMSO vehicle control (e.g., 0.1% v/v) must be included [73].
- Thapsigargin: Dilute the stock solution in complete medium to a final concentration typically between 0.1 and 1 µM, with a corresponding vehicle control [19].
- DTT: Dilute the stock solution in complete medium to a final concentration typically between 1 and 5 mM. A vehicle control with equivalent dilution is recommended.
- Treatment:
- Aspirate the old medium from the cells.
- Gently add the pre-warmed treatment solutions or vehicle control to the cells.
- Incubate the cells for the desired duration at 37°C and 5% CO₂. Treatment times can vary significantly based on the compound and cell type:
- Termination and Analysis: After treatment, the cells or culture supernatants can be harvested for downstream analysis.
Assessment of UPR Activation: Key Methodologies
Confirming UPR activation is a critical step following ER stress induction. Below are key methodologies for monitoring the different UPR arms.
1. XBP1 Splicing Assay (IRE1 Pathway) [58] The unconventional splicing of XBP1 mRNA by activated IRE1 is a hallmark of the UPR.
- Procedure:
- Extract total RNA from treated and control cells.
- Perform reverse transcription (RT) to generate cDNA.
- Amplify the XBP1 region by PCR using gene-specific primers.
- Human XBP1 primers:
- Forward: 5'- TTACGAGAGAAAACTCATGGCC -3'
- Reverse: 5'- GGGTCCAAGTTGTCCAGAATGC -3'
- Human XBP1 primers:
- Resolve the PCR products by agarose gel electrophoresis (e.g., 2.5-3%).
- Interpretation: The unspliced XBP1 (XBP1u) product is 289 bp, while the spliced product (XBP1s) is 263 bp. Activation of IRE1 is indicated by the appearance of the lower band (XBP1s).
2. Immunoblotting for Key UPR Markers Western blot analysis allows for the detection of protein-level changes in UPR components.
- Recommended Targets:
- PERK Pathway: Phospho-eIF2α (Ser51), total eIF2α, ATF4, and CHOP. CHOP induction is a strong indicator of prolonged ER stress [58] [3].
- ATF6 Pathway: Cleavage of full-length ATF6 (90 kDa) to its active cytosolic fragment (ATF6f, ~50 kDa) [58].
- General ER Stress: Upregulation of the chaperone BiP/GRP78 [3].
- Note: Detecting phospho-PERK or cleaved ATF6 can be challenging due to low expression levels and antibody availability. Downstream targets like CHOP and BiP often provide more robust signals [58].
3. Measurement of Cytosolic and ER Calcium Levels This is particularly relevant for thapsigargin experiments.
- Procedure: Use fluorescent, ratiometric calcium indicators (e.g., Fura-2, Fluo-4) according to manufacturer protocols.
- Interpretation: Thapsigargin treatment typically causes a rapid and sustained increase in cytosolic calcium and a corresponding decrease in ER luminal calcium.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for ER Stress Research
| Reagent / Assay | Function in ER Stress Research |
|---|---|
| Tunicamycin | Tool for inducing ER stress via inhibition of protein N-linked glycosylation [72] [73]. |
| Thapsigargin | Tool for inducing ER stress via irreversible inhibition of the SERCA pump and disruption of calcium homeostasis [75] [76]. |
| Dithiothreitol (DTT) | Tool for inducing ER stress by reducing disulfide bonds in the ER, preventing proper protein folding [77]. |
| XBP1 Splicing PCR | A definitive assay for detecting the activation of the IRE1 arm of the UPR [58]. |
| Phospho-eIF2α Antibody | Immunoblot reagent for monitoring the activation of the PERK pathway [58] [3]. |
| CHOP Antibody | Immunoblot reagent for detecting a key transcription factor involved in ER stress-induced apoptosis [58]. Validated antibodies are critical due to reported specificity issues [58]. |
| BiP/GRP78 Antibody | Immunoblot reagent for detecting a central ER chaperone whose expression is upregulated during the UPR [3]. |
Tunicamycin, thapsigargin, and DTT are fundamental pharmacological tools for dissecting the complex biology of ER stress and the UPR. While all three agents ultimately lead to the accumulation of unfolded proteins and UPR activation, their distinct mechanisms—inhibiting glycosylation, depleting calcium, or reducing disulfide bonds—make them uniquely suited for different experimental questions. A thorough understanding of their specific modes of action, optimal dosing, and the appropriate downstream validation assays is essential for designing rigorous experiments. The continued use of these inducers, particularly in combination with modern genetic and biochemical techniques, will undoubtedly yield further insights into the role of ER proteostasis in health and disease, paving the way for novel therapeutic interventions.
The endoplasmic reticulum (ER) serves as a crucial cellular organelle responsible for protein synthesis, folding, and post-translational modifications. The accumulation of unfolded or misfolded proteins within the ER lumen triggers a condition known as ER stress, which activates an adaptive signaling network termed the unfolded protein response (UPR) [78] [3]. The UPR is mediated through three principal ER-transmembrane sensors: inositol-requiring enzyme 1 (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [79] [19]. Under homeostatic conditions, these sensors remain inactive through association with the chaperone protein BiP (GRP78). However, upon ER stress, BiP dissociates to bind misfolded proteins, leading to the activation of these sensors and initiation of downstream signaling cascades that collectively aim to restore proteostasis [78] [3].
In pathological conditions, particularly cancer, UPR signaling is frequently dysregulated, with tumor cells exploiting the adaptive aspects of the UPR to survive in hostile microenvironments characterized by hypoxia, nutrient deprivation, and oxidative stress [79] [80]. This reprogramming enables cancer cells to evade apoptosis, develop therapeutic resistance, and promote angiogenesis and metastasis. Consequently, targeted inhibition of specific UPR pathways has emerged as a promising therapeutic strategy for cancer treatment [81] [80]. This review provides a comprehensive technical overview of preclinical UPR inhibitors, with particular emphasis on STF-083010 (IRE1α inhibitor) and GSK2606414 (PERK inhibitor), detailing their mechanisms of action, experimental applications, and therapeutic potential.
UPR Signaling Pathways and Molecular Mechanisms
The Three Arms of the UPR
IRE1α-XBP1 Pathway: IRE1α is a type I transmembrane protein possessing both kinase and endoribonuclease activities. Upon ER stress, IRE1α oligomerizes and autophosphorylates, activating its RNase domain [81] [79]. The primary substrate of IRE1α's RNase activity is XBP1 mRNA, which undergoes unconventional splicing to produce a potent transcription factor (XBP1s) that upregulates genes involved in ER-associated degradation (ERAD), lipid biosynthesis, and chaperone production [81] [3]. Additionally, activated IRE1α can initiate Regulated IRE1-Dependent Decay (RIDD) of ER-localized mRNAs, reducing the protein-folding load within the ER [81].
PERK-eIF2α Pathway: PERK is similarly activated through oligomerization and autophosphorylation following BiP dissociation. Active PERK phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α), leading to global translational attenuation while selectively promoting the translation of specific mRNAs, notably that of ATF4 [79] [78]. ATF4 then transcriptionally upregulates genes involved in amino acid metabolism, antioxidant responses, and apoptosis. Under prolonged ER stress, ATF4 induces expression of the pro-apoptotic transcription factor CHOP (C/EBP homologous protein), which promotes cell death by regulating Bcl-2 family proteins and oxidative stress [79] [3].
ATF6 Pathway: ATF6 is a type II transmembrane protein that translocates to the Golgi apparatus upon ER stress. In the Golgi, it undergoes proteolytic cleavage by site-1 protease (S1P) and site-2 protease (S2P), releasing its cytosolic domain (ATF6f) [78]. This fragment functions as a transcription factor that migrates to the nucleus to induce expression of ER chaperones and components of the ERAD machinery, thereby enhancing the ER's protein-folding capacity [79] [3].
The following diagram illustrates the core UPR signaling pathways and their modulation by the inhibitors discussed in this review:
The Dual Nature of the UPR in Cell Fate Determination
The UPR exhibits a dualistic nature in determining cellular outcomes. Initially, its signaling is predominantly adaptive and pro-survival, aiming to restore ER homeostasis through multiple mechanisms: (1) transient attenuation of global protein translation to reduce the incoming protein-folding load; (2) transcriptional upregulation of ER chaperones and folding enzymes to enhance folding capacity; and (3) enhancement of ERAD to clear misfolded proteins [79] [78] [3]. However, when ER stress is severe or prolonged, the UPR transitions to a pro-apoptotic signaling mode through several mechanisms: IRE1α-mediated activation of JNK via TRAF2, PERK-ATF4-driven expression of CHOP, and calcium-mediated apoptosis signaling [78] [3]. This delicate balance between pro-survival and pro-apoptotic signaling makes the UPR an attractive therapeutic target, particularly in cancer cells that experience chronic ER stress due to their rapid proliferation and hostile microenvironmental conditions [80].
Preclinical UPR Inhibitors: Mechanism and Application
IRE1α Pathway Inhibitors
STF-083010 is a specific inhibitor of IRE1α's RNase activity. It acts through a mechanism that involves the cleavage of STF-083010 by IRE1α's RNase domain, resulting in covalent modification and irreversible inhibition of the enzyme [81]. This inhibition prevents XBP1 mRNA splicing and the production of the active XBP1s transcription factor, thereby disrupting a crucial adaptive pathway in the UPR.
In preclinical studies, STF-083010 has demonstrated significant antitumor effects. In hepatocellular carcinoma (HCC) models, STF-083010 effectively blocked XBP1 splicing, resulting in reduced tumor cell migration by 60% and decreased lung metastases by 45% [82]. The inhibitor also exhibited potent activity in models of obesity-induced insulin resistance, where its administration significantly ameliorated metabolic parameters by reducing the accumulation of pro-inflammatory adipose tissue macrophages, specifically the 'M1-like' CD11c+ and metabolically activated CD9+ subsets [19] [2]. Furthermore, in organic dust-induced lung inflammation models, STF-083010 suppressed inflammatory mediator production by inhibiting NF-κB-p65, Stat3, Jun, and MAPK 8/9 phosphorylation [83].
Additional IRE1α inhibitors with distinct mechanisms of action have also been developed:
- KIRA6: A kinase-inhibiting RNase attenuator that allosterically inhibits IRE1α's RNase activity by preventing its oligomerization [83].
- APY29: Functions as an ATP-competitive inhibitor of IRE1α's kinase activity, which indirectly suppresses its RNase function [83].
- MKC-3946: Another RNase inhibitor that has shown efficacy in multiple myeloma models by enhancing sensitivity to proteasome inhibitors [81].
PERK Pathway Inhibitors
GSK2606414 is a potent, ATP-competitive inhibitor of PERK's kinase activity. It effectively blocks PERK autophosphorylation, thereby preventing eIF2α phosphorylation and subsequent ATF4 translation [81] [82]. This inhibition disrupts the adaptive PERK signaling pathway, particularly under conditions of ER stress.
In hepatocellular carcinoma studies, GSK2606414 demonstrated remarkable efficacy when combined with sorafenib, reducing HCC cell viability with an IC50 decreased by 40% and inhibiting tumor growth by 55% in vivo [82]. These effects correlated with a significant 60% reduction in ATF4 protein levels, confirming effective target engagement. The compound has also shown promising results in neurodegenerative disease models, though its clinical translation has been hampered by significant on-target toxicity in pancreatic tissues, leading to hyperglycemia in animal models [81].
Alternative PERK-targeting agents include:
- GSK2656157: A derivative of GSK2606414 with improved pharmacokinetic properties, though it still exhibits similar pancreatic toxicity concerns [81].
- ISRIB: An interesting alternative that acts downstream of PERK by reversing the effects of eIF2α phosphorylation, thereby restoring protein translation without directly inhibiting PERK itself [81].
The following table summarizes key preclinical data for these primary UPR inhibitors:
Table 1: Preclinical Profile of Selective UPR Inhibitors
| Inhibitor | Molecular Target | Mechanism of Action | Key Preclinical Findings | Therapeutic Contexts Studied |
|---|---|---|---|---|
| STF-083010 | IRE1α RNase domain | Irreversible, covalent inhibition via cleavage-mediated modification | • Blocked XBP1 splicing, reducing tumor migration by 60% and lung metastases by 45% in HCC models [82]• Reduced pro-inflammatory macrophage accumulation in obesity models [19]• Suppressed organic dust-induced lung inflammation [83] | Hepatocellular carcinoma, obesity-related inflammation, lung inflammation |
| GSK2606414 | PERK kinase domain | ATP-competitive inhibition preventing autophosphorylation | • Combined with sorafenib, reduced HCC cell IC50 by 40% and tumor growth by 55% in vivo [82]• Decreased ATF4 protein levels by 60% [82]• Demonstrated on-target pancreatic toxicity (hyperglycemia) [81] | Hepatocellular carcinoma, various cancers, neurodegenerative diseases |
Experimental Data and Efficacy Metrics
Quantitative assessment of UPR inhibitors in preclinical models provides critical insights into their therapeutic potential and mechanisms of action. The following table compiles key experimental data from published studies:
Table 2: Quantitative Efficacy Data of UPR Inhibitors in Preclinical Models
| Compound | Experimental Model | Treatment Conditions | Key Outcomes | Molecular Changes |
|---|---|---|---|---|
| STF-083010 | HCC models (in vivo) | Monotherapy | • 60% reduction in tumor cell migration• 45% decrease in lung metastases [82] | • 50% reduction in sXBP1 levels [82] |
| GSK2606414 + Sorafenib | HepG2 xenograft (in vivo) | Combination therapy | • 55±7% tumor growth reduction• 40% decrease in IC50 for sorafenib [82] | • 60% reduction in ATF4 protein [82] |
| Tunicamycin (ER stress inducer) | Primary HCC cells | Monotherapy | 60±4% apoptosis induction at 48h [82] | • 3.5-fold increase in CHOP expression [82] |
Research Reagent Solutions: Essential Materials for UPR Studies
Table 3: Key Research Reagents for Investigating UPR Pathways
| Reagent/Category | Specific Examples | Primary Research Application | Key Functions & Mechanisms |
|---|---|---|---|
| UPR Inducers | Tunicamycin, Thapsigargin, Bortezomib, 2-Deoxyglucose | Experimental induction of ER stress | • Tunicamycin inhibits N-linked glycosylation [19]• Thapsigargin disrupts ER calcium homeostasis [19]• Bortezomib inhibits proteasomal degradation [84]• 2-Deoxyglucose inhibits glycosylation (reversible with mannose) [84] |
| IRE1α Pathway Inhibitors | STF-083010, KIRA6, APY29, MKC-3946 | Selective inhibition of IRE1α signaling | • STF-083010: Covalent RNase inhibitor [81] [83]• KIRA6: Kinase-targeting allosteric RNase attenuator [83]• APY29: ATP-competitive kinase inhibitor [83] |
| PERK Pathway Inhibitors | GSK2606414, GSK2656157, ISRIB | Selective inhibition of PERK signaling | • GSK2606414: ATP-competitive PERK kinase inhibitor [81] [82]• ISRIB: Reverses p-eIF2α effects downstream of PERK [81] |
| Protein Synthesis Inhibitors | Moxetumomab pasudotox, Cycloheximide, Puromycin | Arrest of protein translation to block UPR counter-regulation | • Moxetumomab pasudotox: Immunotoxin targeting EF2 [84]• Cycloheximide: Inhibits ribosomal translocation [84] |
| Key Antibodies for UPR Detection | Anti-BiP/GRP78, Anti-p-PERK, Anti-XBP1s, Anti-CHOP | Detection and quantification of UPR activation | • p-PERK: Marker of PERK activation [82]• XBP1s: Indicator of IRE1α activation [82]• CHOP: Marker of pro-apoptotic UPR signaling [82] |
Experimental Protocols for UPR Inhibitor Evaluation
In Vitro Assessment of UPR Inhibitor Efficacy
Cell Viability and Synergy Analysis:
- Cell Culture and Treatment: Maintain cancer cell lines (e.g., HepG2 for HCC, JeKo-1 for mantle cell lymphoma) in appropriate media. Plate cells at optimized densities (typically 5,000-10,000 cells/well in 96-well plates) and allow attachment for 24 hours [84].
- Drug Treatment: Prepare serial dilutions of UPR inhibitors (STF-083010, GSK2606414) alone and in combination with chemotherapeutic agents (e.g., sorafenib for HCC). Include UPR inducers (tunicamycin, thapsigargin) as positive controls for ER stress induction.
- Viability Assessment: After 48-72 hours of treatment, measure cell viability using standardized assays (MTT, CellTiter-Glo). Perform triplicate measurements for each condition.
- Synergy Calculation: Analyze combination effects using Bliss independence model [84]. Calculate fold-change of activity as IC50[A]/IC50[A+B], where values >1 indicate synergy, =1 indicate additivity, and <1 indicate antagonism.
Molecular Validation of UPR Inhibition:
- Western Blot Analysis: Harvest treated cells and prepare protein lysates. Separate proteins by SDS-PAGE and transfer to membranes. Probe with antibodies against:
- XBP1 Splicing Assay: Extract total RNA from treated cells and perform RT-PCR using XBP1-specific primers. Analyze PCR products by agarose gel electrophoresis; unspliced XBP1 yields a 289bp product, while spliced XBP1 yields a 263bp product [84]. Calculate the ratio of XBP1s/XBP1u as a quantitative measure of IRE1α activity.
- Apoptosis Assessment: Evaluate mitochondrial membrane potential using JC-1 dye (shift from red to green fluorescence indicates depolarization). Measure caspase activation through Western blotting for cleaved caspase-3 or fluorogenic caspase activity assays [84].
In Vivo Evaluation of UPR Inhibitors
Animal Model Establishment:
- Xenograft Models: Subcutaneously implant human cancer cells (e.g., HepG2 for HCC) into immunodeficient mice. Allow tumors to reach 100-150mm³ before initiating treatment [82].
- Treatment Protocol: Randomize animals into treatment groups (typically n=5-10 per group). Administer:
- UPR inhibitors alone (e.g., GSK2606414 at 50-100 mg/kg daily by oral gavage)
- Standard chemotherapy alone (e.g., sorafenib for HCC models)
- Combination therapy
- Vehicle control
- Tumor Monitoring: Measure tumor dimensions 2-3 times weekly using calipers. Calculate tumor volume using the formula: V = (length × width²)/2 [82].
- Endpoint Analysis: After 3-4 weeks of treatment, harvest tumors for:
- Weight and volume measurements
- Immunohistochemical analysis of UPR markers (p-PERK, XBP1s, CHOP)
- TUNEL staining for apoptosis assessment
- Microvessel density analysis for angiogenesis evaluation [82]
Pharmacodynamic and Toxicity Assessment:
- Target Engagement Validation: Analyze tumor lysates by Western blotting to confirm reduction in phosphorylated PERK and XBP1s in respective inhibitor treatment groups [82].
- Toxicity Monitoring: Record animal body weight biweekly as an overall health indicator. Collect blood samples for serum chemistry analysis (pancreatic enzymes, liver function tests) to assess specific organ toxicity, particularly pancreatic damage for PERK inhibitors [81].
Pharmacological targeting of UPR signaling represents a promising therapeutic strategy, particularly for cancers characterized by elevated ER stress and adaptive UPR activation. Preclinical inhibitors such as STF-083010 (targeting IRE1α) and GSK2606414 (targeting PERK) have demonstrated significant efficacy in various cancer models, both as monotherapies and in combination with standard chemotherapeutic agents [82] [84]. The experimental protocols outlined provide a standardized framework for evaluating these compounds and novel UPR-targeting agents in preclinical settings.
Future directions in UPR inhibitor development should focus on several critical areas: (1) improving the therapeutic window of UPR inhibitors to minimize on-target toxicities, particularly pancreatic toxicity associated with PERK inhibition; (2) developing biomarker strategies to identify patient populations most likely to benefit from UPR-targeted therapies; and (3) exploring innovative combination regimens that leverage UPR inhibition to overcome resistance to conventional therapies [81] [80]. As our understanding of UPR signaling in pathophysiology continues to evolve, so too will opportunities for therapeutic intervention targeting this critical cellular stress response pathway.
The endoplasmic reticulum (ER) serves as a central hub for cellular biosynthesis, orchestrating the synthesis, folding, and modification of secretory and transmembrane proteins. Approximately one-third of cellular proteins are processed within the ER, where they undergo proper folding and assembly before being transported through the secretory pathway [85]. In the demanding context of cancer, rapidly proliferating tumor cells experience substantial metabolic and environmental stress, leading to an accumulation of misfolded or unfolded proteins within the ER—a condition termed ER stress (ERS) [86]. To restore ER proteostasis and survive under these adverse conditions, cells activate a complex network of adaptive signaling pathways collectively known as the unfolded protein response (UPR) [87].
The UPR is initiated by three ER-resident transmembrane sensors: inositol-requiring enzyme 1α (IRE1α), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [85]. Under normal physiological conditions, these sensors are maintained in an inactive state through association with the chaperone protein GRP78 (also known as BiP). However, the accumulation of misfolded proteins during ER stress triggers GRP78 dissociation, leading to sequential activation of these sensors and their downstream signaling cascades [88]. Cancer cells of various types, including breast, liver, pancreatic, and lung cancers, have been shown to exhibit high levels of UPR activation, which enhances their ability to adapt to unfavorable environments, resist therapeutic interventions, and promote malignant progression [85]. This dependency on UPR signaling creates a therapeutic vulnerability that can be exploited for selective tumor cell elimination.
Molecular Mechanisms of UPR Signaling in Malignancy
IRE1α-XBP1 Signaling Axis
IRE1α, the most conserved UPR sensor, is a type I transmembrane protein possessing both serine/threonine kinase and endoribonuclease activities in its cytoplasmic domain [85]. Upon ER stress, IRE1α dissociates from GRP78, leading to its oligomerization, autophosphorylation, and activation of its RNase function [86]. The primary substrate of IRE1α RNase activity is the mRNA encoding X-box binding protein 1 (XBP1). Activated IRE1α catalyzes the unconventional splicing of a 26-nucleotide intron from XBP1 mRNA, resulting in a translational frameshift that produces the potent transcription factor XBP1s (spliced form) [85]. XBP1s then translocates to the nucleus and upregulates a diverse set of target genes involved in ER biogenesis, protein folding, quality control, and ER-associated degradation (ERAD), thereby enhancing the ER's protein-processing capacity [85].
In cancer, the IRE1α-XBP1 axis is hijacked to support tumor progression across multiple malignancies. In triple-negative breast cancer (TNBC), XBP1 is hyperactivated and promotes tumor progression, correlating with poorer relapse-free survival [88]. The IRE1α-XBP1 pathway significantly accentuates MYC-driven tumorigenesis in breast cancer and urothelial carcinomas [86]. Furthermore, extracellular vesicles from cancer cells exploit the IRE1α pathway to instigate oncogenic transformation in bladder cancer [86]. Beyond cell-autonomous effects, IRE1α-XBP1 signaling governs cancer stemness and reprograms lipid metabolism in tumor-infiltrating immune cells, representing a compelling immunotherapeutic target [88].
PERK-eIF2α-ATF4 Signaling Axis
PERK, a type I transmembrane kinase, follows a similar activation mechanism to IRE1α. Upon ER stress-induced GRP78 dissociation, PERK undergoes oligomerization and autophosphorylation, leading to kinase activation [85]. The primary substrate of PERK is the α-subunit of eukaryotic initiation factor 2 (eIF2α). Phosphorylation of eIF2α at serine 51 inhibits guanine nucleotide exchange, thereby attenuating global protein translation to reduce the protein-folding burden on the stressed ER [85]. Paradoxically, this translational attenuation enables selective translation of specific mRNAs, including that of activating transcription factor 4 (ATF4) [85]. ATF4 then orchestrates the expression of genes involved in amino acid metabolism, antioxidant responses, and autophagy. Under prolonged ER stress, ATF4 induces the expression of the transcription factor C/EBP homologous protein (CHOP), which plays a crucial role in directing the UPR toward pro-apoptotic outcomes [85].
The PERK pathway exhibits dual roles in cancer biology, functioning as both a pro-survival and pro-apoptotic signal depending on context and duration. PERK activation helps tumor cells survive under hypoxic conditions and enhances their migratory and metastatic potential [88]. For instance, PERK activation induces the metastasis-associated gene LAMP3, linking UPR activation to increased metastatic potential [88]. Additionally, PERK activity enables cancer cells to resist oxidative DNA damage, further supporting tumor growth [88]. However, in contrast to its pro-tumorigenic functions, the PERK pathway exerts tumor-suppressive effects in certain contexts, particularly in breast cancer, where PERK activation significantly inhibits tumor proliferation [86].
ATF6 Signaling Pathway
ATF6 exists as a type II transmembrane protein anchored to the ER and is characterized by its transient activity with a notably short half-life of approximately two hours [86]. Unlike PERK and IRE1α, ATF6 activation occurs through translocation to the Golgi apparatus. Following ER stress, full-length ATF6 traffics to the Golgi, where it undergoes proteolytic cleavage by site-1 protease (S1P) and site-2 protease (S2P), releasing its cytosolic domain [86]. This active ATF6 fragment then translocates to the nucleus and functions as a transcription factor, regulating numerous UPR target genes including GRP78, GRP94, ERAD components, XBP1, and CHOP [86]. Additionally, ATF6 can form heterodimers with XBP1, enhancing ERAD with increased affinity for UPR elements compared to XBP1 homodimers [86].
In cancer, ATF6 emerges as a key downstream effector with significant roles in oncological biology. Elevated ATF6α is closely associated with prostate cancer progression, where it regulates arachidonic acid metabolism via the ATF6α-PLA2G4A signaling pathway [86]. ATF6 also promotes malignancy through a reciprocal negative feedback mechanism with PTEN and maintains BRCA-1 expression, enabling colon cancer cells to evade DNA damage and enhance viability [86]. In colorectal cancer, phosphorylated ATF6 promotes tumor development by inducing gut microbiome dysbiosis and activating inflammatory pathways [86].
Therapeutic Targeting of UPR Pathways in Cancer
Pharmacological Inhibitors of UPR Components
The heightened dependence of cancer cells on UPR pathways for survival under stress has motivated the development of pharmacological agents targeting specific UPR components. These inhibitors aim to disrupt the adaptive UPR, pushing cancer cells from pro-survival signaling toward apoptotic elimination. Both natural and synthetic compounds have been investigated that either induce persistent ERS by selectively blocking ER Ca2+ pumps (SERCA) to disrupt ER Ca2+ homeostasis or alter the activity of UPR chaperones and sensors to impair protein degradation signaling [87].
Table 1: Pharmacological Agents Targeting UPR Pathways in Cancer
| Target | Therapeutic Agent | Mechanism of Action | Experimental Context | Therapeutic Effect |
|---|---|---|---|---|
| GRP78 | Various inhibitors | Disrupts chaperone function, preventing sensor inactivation | Preclinical studies | Sensitizes cancer cells to conventional chemotherapy [87] |
| IRE1α | MKC-8866, B-I09 | Inhibits RNase activity, blocking XBP1 splicing | Preclinical models | Induces apoptosis in AML; shows efficacy in combination therapy [88] |
| PERK | AMG 44 | Inhibits kinase activity, preventing eIF2α phosphorylation | Preclinical models | Reduces IRE1 expression; shows synergy in combination approaches [88] |
| ATF6 | Ceapins | Selective inhibition of ATF6 signaling | Experimental settings | Impairs ER stress adaptation in cancer cells [87] |
| SERCA | Thapsigargin analogs | Disrupts ER calcium homeostasis, inducing persistent ER stress | Preclinical studies | Triggers irreversible ER stress, leading to cell death [87] |
Experimental Protocols for UPR Modulation Studies
Protocol 1: Assessing XBP1 Splicing as a Measure of IRE1α Activity
XBP1 splicing serves as a definitive indicator of IRE1α RNase activity and is widely used to monitor UPR activation in experimental systems. To assess XBP1 splicing, researchers typically isolate total RNA from treated cells using standard TRIzol or column-based methods. Reverse transcription is performed using gene-specific primers or random hexamers, followed by PCR amplification with primers flanking the IRE1α cleavage site in XBP1 mRNA. The PCR products are then separated by agarose gel electrophoresis (typically 2.5-3%), which distinguishes the unspliced (XBP1u, ~480 bp) and spliced (XBP1s, ~456 bp) variants due to the 26-nucleotide excision [85]. For quantitative assessment, real-time PCR assays with specific probes for XBP1u and XBP1s can be employed. This protocol is particularly useful for evaluating IRE1α inhibitors such as MKC-8866 and B-I09, where reduction in XBP1s levels indicates effective target engagement [88].
Protocol 2: Monitoring elF2α Phosphorylation as a Readout of PERK Activity
PERK activation is routinely assessed by measuring phosphorylation of its primary substrate, eIF2α, at serine 51. This analysis typically involves protein extraction from treated cells using RIPA buffer supplemented with phosphatase and protease inhibitors. Protein concentrations are determined by BCA or Bradford assay, and equal amounts of protein are separated by SDS-PAGE (10-12% gels) before transfer to PVDF or nitrocellulose membranes. Immunoblotting is performed using phospho-specific eIF2α (Ser51) antibodies, followed by stripping and reprobing with total eIF2α antibodies to normalize for protein loading. Densitometric analysis of band intensities provides quantitative assessment of PERK activation. This method is essential for evaluating PERK pathway modulators and understanding the temporal dynamics of PERK signaling in response to ER stress-inducing agents [85].
Protocol 3: Evaluating Cytotoxicity and Synergy in Combination Therapies
Given that UPR-targeting agents are primarily investigated in combination approaches, robust assessment of cytotoxicity and drug synergy is crucial. Researchers typically employ cell viability assays (MTT, WST-1, or CellTiter-Glo) following exposure to UPR inhibitors alone and in combination with standard chemotherapeutics. For synergy analysis, combination indices are calculated using methods described by Chou-Talalay, where values less than 1 indicate synergy. Additionally, apoptosis assays using Annexin V/propidium iodide staining with flow cytometry provide mechanistic insight into cell death pathways engaged by UPR disruption. These protocols are fundamental for establishing the therapeutic potential of UPR modulation and guiding clinical translation of combination strategies [87] [88].
UPR Modulation in the Tumor Microenvironment and Therapeutic Implications
UPR-Mediated Immune Modulation
The therapeutic potential of UPR modulation extends beyond cancer cell-autonomous effects to encompass significant impacts on the tumor immune microenvironment. Immune cells within the TME also experience severe ER stress, and UPR activation in these cells leads to a suppressive immune microenvironment, converting anticancer responses into procancer effects [85]. The IRE1α-XBP1 axis governs the lipid metabolism of tumor-infiltrating dendritic cells and T cells, rendering it a compelling immunotherapeutic target [88]. CHOP signaling and myeloid IRE1α signaling can dampen antitumor immunity, providing a rationale to pair UPR modulation with immunotherapy [88]. UPR activation in immune cells promotes cancer immune editing and escape through multiple mechanisms, including impairment of antigen-presenting cell function, activation of myeloid-derived suppressor cells (MDSCs), and promotion of T cell exhaustion [89].
Table 2: UPR Modulation in Cancer Clinical Development
| Therapeutic Agent | Molecular Target | Cancer Type | Development Stage | Key Findings |
|---|---|---|---|---|
| Rezatapopt | TP53 Y220C mutation | Advanced solid tumors (ovarian, lung, breast, etc.) | Phase 2 trial | 34% ORR in evaluable patients; 46% ORR in ovarian cancer; median DOR 7.6 months [90] |
| UPR modulators + Immunotherapy | Multiple UPR pathways | Gynecologic cancers, other solid tumors | Preclinical and early clinical | Synergy with PD-1 inhibitors; enhanced antitumor immunity [88] |
| UPR modulators + Chemotherapy | GRP78, IRE1α, PERK | Ovarian cancer, AML, others | Preclinical and early clinical | Reversal of chemoresistance; enhanced sensitivity to platinum agents [87] [88] |
Biomarker Development and Patient Stratification
The successful clinical translation of UPR-targeting therapies requires robust biomarker development for patient stratification and treatment response monitoring. In epithelial ovarian cancer, GRP78, ATF6, and PERK overexpression correlates with inferior survival and chemoresistance, supporting their utility as biomarkers and therapeutic targets [88]. Similarly, in acute myeloid leukemia (AML), a UPR-related gene signature (URGsig) has been developed that enables stratification of patients into distinct risk subgroups with significant differences in survival outcomes, tumor immune landscape, and therapy sensitivity [89]. These UPR-based prognostic signatures demonstrate the clinical applicability of UPR biomarkers and provide a framework for personalized treatment approaches. Somatic mutations in UPR-related genes are prevalent across gynecological malignancies, with IRE1α mutations often abrogating RIDD activity while retaining XBP1 splicing, conferring survival advantages that may inform targeted therapy selection [88].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for UPR Studies
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| UPR Activators | Tunicamycin, Thapsigargin, Brefeldin A | Induction of ER stress as positive control | Concentrations and duration must be optimized for each cell type |
| IRE1α Inhibitors | MKC-8866, B-I09, 4μ8C | Specific inhibition of IRE1α RNase activity | Monitor via XBP1 splicing assays; off-target effects on other RNases possible |
| PERK Inhibitors | GSK2606414, AMG 44 | Selective PERK kinase inhibition | Assess via eIF2α phosphorylation immunoblotting; can activate compensatory IRE1α |
| ATF6 Inhibitors | Ceapins | Selective blockade of ATF6 activation | Evaluate via GRP78 promoter reporter assays |
| GRP78 Inhibitors | HA15, Viridin analogs | Disruption of GRP78 chaperone function | Can induce protective autophagy; often used in combination approaches |
| Antibodies | Phospho-eIF2α (Ser51), Total eIF2α, XBP1s, CHOP, GRP78 | Protein detection and pathway activation assessment | Always validate antibodies for specific applications; use phospho-specific antibodies for activation states |
| Cell Lines | MCF-7 (breast), PC-3 (prostate), A549 (lung), OVCAR-3 (ovarian) | Model systems for UPR studies | Select lines with basal UPR activation; authenticate regularly |
| Reporters | GRP78 promoter-luciferase, XBP1 splicing reporter | Real-time monitoring of UPR activation | Transient vs. stable transfection affects response dynamics |
The strategic modulation of UPR pathways represents a promising approach for selective tumor cell elimination that leverages the inherent metabolic vulnerabilities of cancer cells. The complex interplay between the three UPR arms—IRE1α, PERK, and ATF6—creates both challenges and opportunities for therapeutic intervention. While preclinical evidence supporting UPR modulation as an anticancer strategy is compelling, clinical translation remains limited, with inconsistent results and challenges related to toxicity, bioavailability, and therapeutic specificity [87]. Future research priorities include validating UPR-based prognostic signatures, defining context-specific vulnerabilities, and executing biomarker-driven clinical trials that combine UPR-targeted agents with standard chemotherapy, PARP inhibition, and immunotherapy to overcome resistance and improve patient outcomes [88]. The development of more selective UPR-targeting agents with improved stability and reduced toxicity, coupled with advanced delivery systems to enhance therapeutic specificity, represents a critical frontier in realizing the potential of UPR modulation for cancer therapy.
The unfolded protein response (UPR), a crucial cellular stress signaling network, represents a promising therapeutic target for mitigating proteotoxicity in neurodegenerative diseases. This whitepaper delineates the molecular mechanisms of UPR signaling branches—PERK, IRE1α, and ATF6—and their dual roles in neuronal survival and apoptosis under chronic ER stress conditions. We present quantitative analyses of UPR modulation outcomes, detailed experimental methodologies for target validation, and visualize critical signaling pathways and therapeutic strategies. Furthermore, we catalog essential research tools and reagents advancing drug discovery. The synthesized evidence underscores the potential of precision UPR modulation to counteract protein aggregation in conditions including Alzheimer's disease, Parkinson's disease, and other proteopathies, thereby providing a framework for developing novel neuroprotective interventions.
The endoplasmic reticulum (ER) serves as the primary intracellular organelle for protein synthesis, folding, and quality control. Maintaining ER homeostasis, or "proteostasis," is critical for cellular function, particularly in long-lived, post-mitotic neurons [3]. Numerous pathological insults—including oxidative stress, metabolic dysregulation, genetic mutations, and aging—can disrupt ER function, leading to an accumulation of misfolded or unfolded proteins, a condition termed ER stress [91] [3]. To counteract this, cells activate the unfolded protein response (UPR), a sophisticated signaling network orchestrated by three ER-resident transmembrane sensors: PERK (PKR-like ER kinase), IRE1α (inositol-requiring enzyme 1 alpha), and ATF6 (activating transcription factor 6) [2] [3].
The UPR is a quintessential double-edged sword in cellular stress adaptation. Its initial, transient activation is adaptive and pro-survival, aiming to restore proteostasis by reducing global protein synthesis, enhancing the ER's protein-folding capacity, and promoting the degradation of misfolded proteins. However, under severe or prolonged ER stress, the UPR switches to a pro-apoptotic signaling program, eliminating irreversibly damaged cells [91] [3] [92]. In neurodegenerative diseases, characterized by the accumulation of misfolded proteins like amyloid-β and α-synuclein, chronic ER stress and the maladaptive UPR drive neuronal loss [93] [92]. Consequently, understanding and therapeutically targeting the UPR's delicate balance presents a compelling strategy for alleviating proteotoxicity.
Molecular Mechanisms of the UPR
The three UPR sensors are maintained in an inactive state through association with the ER chaperone BiP (GRP78). The accumulation of unfolded proteins recruits BiP away from these sensors, triggering their activation and initiating distinct downstream signaling cascades [3]. Figure 1 illustrates the core UPR signaling pathways and their functional outcomes.
The PERK-eIF2α-ATF4 Pathway
Upon ER stress, PERK dimerizes and autophosphorylates, activating its kinase domain. Its primary substrate is the eukaryotic initiation factor 2α (eIF2α). Phosphorylation of eIF2α (p-eIF2α) globally attenuates cap-dependent protein translation, reducing the incoming protein load on the stressed ER [3] [17]. Paradoxically, this translation inhibition facilitates the selective synthesis of specific mRNAs, notably that of the transcription factor ATF4. ATF4 upregulates genes involved in amino acid metabolism, antioxidant responses, and, critically, the pro-apoptotic factor CHOP (C/EBP-homologous protein). CHOP induces expression of genes that promote oxidative damage and apoptosis, such as ERO1α, making the PERK-CHOP axis a key mediator of ER stress-induced cell death [3] [93].
The IRE1α-XBP1 Pathway
IRE1α, the most conserved UPR sensor, possesses both kinase and endoribonuclease activities. Its activation leads to the unconventional splicing of the mRNA encoding X-box binding protein 1 (XBP1). The spliced XBP1s mRNA is translated into a potent transcription factor that drives the expression of genes involved in ER chaperone production, ER-associated degradation (ERAD), and lipid biosynthesis, collectively expanding the ER's folding and processing capacity [2] [3]. Under sustained stress, IRE1α can also trigger pro-apoptotic signaling through its interaction with TRAF2, leading to the activation of JNK (c-Jun N-terminal kinase) [93].
The ATF6 Pathway
ER stress triggers the translocation of ATF6 to the Golgi apparatus, where it is cleaved by proteases (S1P and S2P). This regulated intramembrane proteolysis releases its cytosolic domain, ATF6f, which functions as a transcription factor. ATF6f migrates to the nucleus to induce genes encoding ER chaperones (e.g., BiP, GRP94) and components of the ERAD machinery, acting in concert with XBP1s to enhance the ER's protein-handling capabilities [2] [3].
Figure 1. Core UPR Signaling Pathways and Functional Outcomes. The accumulation of unfolded proteins in the ER lumen activates three sensors—PERK, IRE1α, and ATF6—triggering adaptive responses. Severe or prolonged stress switches signaling toward pro-apoptotic outcomes through effectors like CHOP and JNK. Abbreviations: p-, phosphorylated; ERAD, ER-associated degradation.
Quantitative Evidence: UPR Modulation Outcomes
Strategic inhibition of specific UPR branches has demonstrated significant neuroprotective effects in preclinical models. The table below summarizes key quantitative findings from recent studies.
Table 1: Quantitative Outcomes of UPR Pathway Modulation in Preclinical Models
| Disease Model | Intervention / Genetic Manipulation | Key Quantitative Outcomes | Primary UPR Branch Targeted | Reference |
|---|---|---|---|---|
| Rotenone-induced PD model (SH-SY5Y cells) | AMG44 (PERK/CHOP inhibitor) | ✓ Significant prevention of ROT-induced viability decrease✓ Reduced apoptosis and necrosis✓ Attenuated ROS generation | PERK-CHOP | [93] |
| Rotenone-induced PD model (SH-SY5Y cells) | JNK V (IRE1/JNK inhibitor) | ✓ Significant prevention of ROT-induced viability decrease✓ Reduced apoptosis and necrosis✓ Attenuated ROS generation | IRE1-JNK | [93] |
| Aβ C. elegans AD model | Lycium barbarum extract (LBE) | ✓ Significant reduction in Aβ aggregation✓ Activation of mtUPR via FSHR-1✓ Improved muscle motility phenotype | mtUPR (Activation) | [94] |
| Diet-induced obesity (Mice) | STF-083010 (IRE1α RNase inhibitor) | ✓ Ameliorated insulin resistance✓ Reduced pro-inflammatory M1 macrophage accumulation✓ Increased thermogenesis | IRE1α-XBP1 | [2] |
| TDP-43 proteinopathy mouse model | AAV8-delivered TDP-43 siRNA (via sEVs) | ✓ Improved motor function✓ Reduced neuronal TDP-43 expression✓ Decreased neuronal loss and gliosis | N/A (Reduces pathological protein load) | [95] |
These findings highlight that targeted pharmacological inhibition of the pro-apoptotic PERK/CHOP and IRE1/JNK arms can preserve neuronal viability, while activating protective mechanisms like the mtUPR can alleviate proteotoxicity.
Experimental Protocols for UPR Research
Protocol: Evaluating UPR Inhibitors in a Cellular PD Model
This methodology is adapted from a study investigating PERK and IRE1 pathway inhibitors in rotenone-treated SH-SY5Y cells [93].
- Cell Culture and Differentiation: Maintain human neuroblastoma SH-SY5Y cells in standard culture medium. Differentiate cells by treatment with 10 µM retinoic acid for 5-7 days to induce a mature neuronal phenotype.
- ER Stress Induction and Inhibitor Treatment:
- Prepare a 1 mM stock solution of rotenone in DMSO and dilute in culture medium to a final working concentration (e.g., 100-500 nM).
- Reconstitute UPR pathway inhibitors: AMG44 (PERK/CHOP inhibitor) and JNK V (IRE1/JNK inhibitor) in DMSO.
- Apply inhibitor compounds (e.g., 1-10 µM) to cells 1-2 hours prior to rotenone exposure.
- Assessment of Cell Viability and Death:
- MTT Assay: After 24 hours of rotenone exposure, add MTT reagent (0.5 mg/mL) to cells and incubate for 4 hours. Dissolve the resulting formazan crystals in DMSO and measure absorbance at 570 nm. Calculate viability relative to untreated controls.
- Flow Cytometry for Apoptosis/Necrosis: Use an Annexin V-FITC/PI dual-staining kit. Resuspend harvested cells in binding buffer, incubate with Annexin V-FITC and PI for 15 minutes in the dark, and analyze by flow cytometry within 1 hour.
- Measurement of Reactive Oxygen Species (ROS):
- Load cells with 10 µM DCFH-DA fluorescent probe for 30 minutes at 37°C.
- Wash cells with PBS and analyze fluorescence intensity immediately using a fluorescence microplate reader (excitation/emission: 485/535 nm) or flow cytometry.
- Gene and Protein Expression Analysis:
- qRT-PCR: Extract total RNA and reverse transcribe to cDNA. Perform qPCR using primers for UPR-related genes (e.g., CHOP, XBP1s, BiP, ATF4). Normalize data to a housekeeping gene (e.g., GAPDH).
- Western Blotting: Lyse cells in RIPA buffer. Separate proteins by SDS-PAGE, transfer to a membrane, and probe with primary antibodies against p-PERK, p-eIF2α, CHOP, XBP1s, p-JNK, and a loading control (e.g., β-actin).
Protocol: Deep Visual Proteomics (DVP) for Spatial UPR Analysis
This cutting-edge protocol, used to map proteotoxicity in human liver tissue, can be adapted to study brain samples from neurodegenerative disease models or patients [96].
- Tissue Preparation: Use formalin-fixed, paraffin-embedded (FFPE) tissue sections (3-10 µm thick) mounted on membrane slides.
- Staining and AI-Guided Cell Classification:
- Stain sections with antibodies against proteins of interest (e.g., pathological protein aggregates like Aβ or p-tau) and a nuclear stain.
- Acquire high-resolution images. Use a trained machine learning algorithm to segment and classify individual cells or regions based on staining intensity and morphological features (e.g., "low," "moderate," or "high aggregate load").
- Laser Microdissection: Using the digital classification map as a guide, laser-capture the predefined cellular regions of interest.
- Sample Processing for Mass Spectrometry:
- Digest microdissected tissue samples with trypsin/Lys-C directly on the membrane.
- Desalt and purify the resulting peptides.
- High-Sensitivity Mass Spectrometry:
- Analyze peptides on a high-performance Orbitrap Astral mass spectrometer.
- Use a data-independent acquisition (DIA) mode for deep proteome coverage from minimal input material.
- Bioinformatic Analysis:
- Match MS/MS spectra to protein sequence databases for identification and quantification.
- Perform pathway enrichment analysis (e.g., for UPR, oxidative stress, peroxisomal pathways) and correlate protein expression levels with pathological load across the different classified cell groups.
Figure 2. Deep Visual Proteomics (DVP) Workflow. This integrated approach combines immunohistochemistry, artificial intelligence, laser microdissection, and high-sensitivity mass spectrometry to achieve spatially resolved, deep proteomic profiling from complex tissues like the brain, enabling the correlation of UPR pathway activation with specific pathological features at single-cell resolution.
Therapeutic Strategies and Research Toolkit
Targeted Therapeutic Approaches
Therapeutic strategies aim to inhibit pro-apoptotic UPR arms or enhance its adaptive, pro-survival functions.
Inhibition of Pro-apoptotic UPR Signaling: Small-molecule inhibitors targeting specific nodes of the PERK/CHOP and IRE1/JNK pathways, such as AMG44 and JNK V, have demonstrated efficacy in protecting dopaminergic neurons in a rotenone-induced model of Parkinson's disease [93]. This approach seeks to suppress the terminal, cytotoxic output of the UPR while preserving its initial adaptive signaling.
Activation of Protective UPR and Related Pathways: Natural products like Lycium barbarum extract (LBE) can activate the mitochondrial UPR (mtUPR), a complementary stress response pathway that maintains mitochondrial proteostasis. In a C. elegans model of Alzheimer's disease, LBE reduced Aβ aggregation and improved phenotypes via mtUPR activation, independently of the ER UPR [94]. This highlights the potential of boosting parallel proteostatic mechanisms.
Reducing the Overall Proteotoxic Load: An alternative strategy involves directly lowering the levels of aggregation-prone proteins. A novel gene therapy strategy used engineered small extracellular vesicles (sEVs) decorated with the RVG peptide to deliver TDP-43 siRNAs across the blood-brain barrier. This approach significantly reduced TDP-43 pathology and improved motor function in a mouse model, demonstrating the utility of tackling the root cause of proteotoxicity [95].
The Scientist's Toolkit: Key Research Reagents
The table below catalogues critical reagents and tools for investigating UPR in neurodegenerative disease models.
Table 2: Essential Research Reagents for UPR Studies
| Reagent / Tool | Function / Target | Application in UPR/Neurodegeneration Research | Example |
|---|---|---|---|
| ER Stress Inducers | Disrupt ER proteostasis | Experimentally induce ER stress to study UPR activation and neuronal vulnerability. | Tunicamycin (N-glycosylation inhibitor), Thapsigargin (SERCA pump inhibitor) [2] [17] |
| UPR Pathway Inhibitors | Specific UPR branches | Mechanistic dissection of UPR contributions and evaluation of therapeutic candidates. | AMG44 (PERK/CHOP inhibitor), STF-083010 (IRE1α RNase inhibitor), JNK V (JNK inhibitor) [2] [93] |
| UPR Reporter Cell Lines | Monitor UPR activation | Real-time, non-invasive tracking of specific UPR branch activity in live cells. | XBP1-splicing reporter (for IRE1 activity), CHOP-promoter luciferase reporter (for PERK pathway) |
| Validated Antibodies | Detect UPR components | Western blot, immunohistochemistry, and immunofluorescence for assessing UPR activation. | Antibodies against p-PERK, p-eIF2α, CHOP, XBP1s, BiP/GRP78 [93] |
| Animal Models | Model proteinopathies | In vivo study of UPR in disease progression and therapy testing. | Aβ-expressing C. elegans (e.g., CL2006), TDP-43 mouse model, Rotenone-induced PD model [93] [94] [95] |
| AAV Vectors / sEV Tools | Gene delivery to CNS | Modulate gene expression (e.g., UPR components, pathogenic proteins) in the brain. | AAV8 for liver-targeting, RVG-Lamp2a for neuron-targeting sEVs [95] |
Targeting the UPR represents a rational and promising frontier for developing disease-modifying therapies for neurodegenerative disorders. The evidence synthesized herein confirms that precise modulation of specific UPR branches—particularly the suppression of the pro-apoptotic PERK/CHOP and IRE1/JNK axes or the potentiation of protective mechanisms like the mtUPR—can effectively alleviate proteotoxicity and confer neuroprotection in preclinical models. The ongoing development of sophisticated research tools, such as Deep Visual Proteomics and novel brain-penetrant delivery systems, will provide unprecedented spatial resolution in understanding UPR pathophysiology and enable more effective therapeutic targeting.
Future efforts must focus on achieving context-dependent UPR modulation, as the response is highly dynamic and can vary by cell type, disease stage, and the nature of the proteotoxic insult. The challenge remains to selectively inhibit the maladaptive, pro-death signals while sparing or enhancing the beneficial, homeostatic functions of the UPR. Combining UPR-targeted agents with other therapeutic modalities, such as those aimed at reducing the production of aggregation-prone proteins or enhancing their clearance, may offer synergistic benefits. As research progresses, targeting the UPR is poised to transition from a compelling concept to a tangible clinical strategy for combating neurodegenerative diseases.
The endoplasmic reticulum (ER) is the largest organelle in mammalian cells, performing essential functions including the synthesis, transport, and folding of proteins, lipid and steroid synthesis, carbohydrate metabolism, and calcium storage [2] [19]. A dysfunction in the ER's protein-folding capacity leads to the accumulation of unfolded or misfolded proteins, a state known as "ER stress" [2]. To counteract this stress, cells activate a sophisticated signaling network known as the Unfolded Protein Response (UPR), which is orchestrated by three ER-transmembrane sensors: Inositol-requiring enzyme 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor-6 (ATF6) [2] [19].
The UPR displays a dual nature in determining cell fate. In its initial, adaptive phase, it aims to restore proteostasis by attenuating global protein translation, upregulating ER chaperones, and enhancing ER-associated degradation (ERAD) of misfolded proteins [2] [91]. However, under severe or prolonged stress conditions, the UPR switches from this pro-survival program to a pro-apoptotic one, triggering cell death [2] [91]. Chronic ER stress and dysregulated UPR are now established as pivotal drivers in the pathogenesis of numerous human diseases, including metabolic conditions like diabetes and obesity, neurodegenerative disorders such as Alzheimer's and Parkinson's disease, autoimmune conditions including rheumatoid arthritis and systemic lupus erythematosus, and cancer [2] [91] [17].
Table 1: Core Components of the Unfolded Protein Response (UPR)
| UPR Arm | ER Sensor | Key Signaling Mechanism | Primary Transcription Factor | Major Functional Outcomes |
|---|---|---|---|---|
| IRE1α Pathway | IRE1α | Autophosphorylation and activation of its RNase activity; splices XBP1 mRNA | XBP1s (spliced XBP1) | Enhances ER folding capacity and ER-associated degradation (ERAD) [2] [91] |
| PERK Pathway | PERK | Phosphorylates eIF2α to attenuate translation; selective translation of ATF4 | ATF4, CHOP | Reduces ER protein load; regulates antioxidant response; can induce apoptosis [2] [91] |
| ATF6 Pathway | ATF6 | Translocates to Golgi for proteolytic cleavage | ATF6f (ATF6 fragment) | Upregulates ER chaperones and components of ERAD pathway [2] [91] |
Within immune cells, the ER serves as a critical nexus for sensing cellular status and orchestrating adaptive responses [2]. The UPR has emerged as a central regulator of immune cell development, activation, differentiation, and effector functions, influencing processes ranging from cytokine production to polarization fate decisions [2] [19] [97]. This technical review examines the cell-type-specific functions of ER stress and UPR signaling across the immune spectrum and explores the emerging therapeutic potential of targeting these pathways in inflammatory diseases.
UPR Signaling in Innate Immune Cell Function
Monocytes and Macrophages
Monocytes circulating in blood defend against pathogens via phagocytosis and can differentiate into macrophages or dendritic cells to support immune responses and tissue repair [2]. In these cells, ER stress directly promotes an inflammatory phenotype. Thapsigargin (Tg)-induced ER stress in monocytes increases mRNA expression of pro-inflammatory cytokines IL-6 and IL-8 and amplifies TNF-α production in response to lipopolysaccharide (LPS) and palmitic acid [2] [19].
Macrophages display remarkable plasticity, with their activation states described as a spectrum between pro-inflammatory 'M1' and anti-inflammatory, tissue-reparative 'M2' phenotypes [2]. UPR signaling directly influences this polarization. In macrophages, saturated fatty acids engage the IRE1α pathway to promote activation of the NLRP3 inflammasome, leading to secretion of IL-1β, a cytokine closely linked to insulin resistance [2] [19]. A key study demonstrated that myeloid-specific deletion of IRE1α protects mice from diet-induced obesity and insulin resistance by promoting a shift from pro-inflammatory M1 to anti-inflammatory M2 macrophage polarization in adipose tissue [2]. Corroborating these findings, pharmacological inhibition of IRE1α's RNase activity with STF-083010 significantly ameliorated insulin resistance and protected against obesity in mice by reducing accumulation of pro-inflammatory adipose tissue macrophages [2] [19].
Beyond IRE1α, other UPR branches contribute to macrophage activation. Deficiency of ATF4, a transcription factor downstream of the PERK pathway, suppresses saturated fatty acid-induced expression of IL-6 in macrophages [2] [19]. Additionally, high-fat diet-induced CHOP expression within adipocytes drives polarization of adipose tissue macrophages toward the pro-inflammatory M1 phenotype, resulting in insulin resistance [2].
Dendritic Cells and Granulocytes
As professional antigen-presenting cells, dendritic cells (DCs) play critical roles in both innate and adaptive immunity by presenting antigens to naïve T cells and producing instructive cytokines such as IL-12 and IL-23 that direct T cell differentiation [2] [19]. In DCs, the ATF6α pathway contributes to the production of critical pro-inflammatory cytokines including IL-12p70 and IL-6 upon activation [2]. Other UPR arms also modulate cytokine production; induction of ER stress with tunicamycin or thapsigargin in pathogen-associated molecular pattern-stimulated DCs markedly enhanced mRNA expression of the pro-inflammatory cytokine IL-23 [2] [19]. The IRE1α pathway was essential for the IL-23 response to the fungal pathogen-associated molecular pattern zymosan, while the PERK pathway was required for the response to bacterial LPS [2].
XBP1s is indispensable for the development and survival of DCs [2]. Recent reports indicate that TRIM29, a member of E3 ubiquitin ligase, promotes PERK-mediated ER stress immune response by inducing SUMOylation and stabilizing PERK [2]. Since TRIM29 negatively regulates innate immune responses against viral infections by inhibiting type I interferon production in DCs and macrophages, the TRIM29-PERK axis represents a potential target for ER stress-associated immune disorders [2].
Granulocytes, including neutrophils, eosinophils, and basophils, play important roles in inflammation through pathogen clearance and immunoregulation [2]. While the search results provide limited specific details on granulocyte UPR signaling, it is noted that neutrophils, as essential first responders in acute inflammation, exacerbate disease through mechanisms that may involve UPR pathways [2].
Table 2: UPR-Mediated Inflammatory Responses in Key Immune Cells
UPR in Adaptive Immunity and Inflammatory Disease Pathogenesis
T Cell and B Cell Regulation
While the provided search results focus more extensively on innate immune cells, they establish that ER stress and UPR are critical regulators of both innate and adaptive immune cells [2] [19]. The ER serves as a essential site not only for protein synthesis but also for integrating environmental signals that influence immune cell fate and function [2]. Dysregulated ER stress responses can profoundly disrupt immune homeostasis and contribute to pathological inflammation and tissue damage across various disease states [2].
UPR in Inflammatory Disease: Celiac Disease Case Study
Celiac disease provides a compelling example of UPR involvement in inflammatory disease pathogenesis. Research demonstrates that gliadin peptides from digested gluten promptly induce ER stress in intestinal epithelial cells, activating all three UPR transcription factors: ATF4, ATF6, and XBP1 [98]. This UPR activation directly contributes to key pathological features of celiac disease, including upregulation of transglutaminase 2 (TG2), dysregulation of CFTR, altered expression of tight junction proteins (increased OCL, decreased CLD-2 and CLD-15), and production of pro-inflammatory cytokines (IFNγ, IL-15, and IL-17A) [98].
Notably, administration of chemical chaperones that buffer ER stress prevented these pathological changes, restored physiological protein expression patterns, and attenuated inflammation in experimental models [98]. These findings highlight the central role of UPR signaling in celiac disease pathogenesis and identify chemical chaperones as potential therapeutic interventions.
Experimental Models and Methodologies for UPR Research
Established Models for UPR Investigation
Researchers employ various well-established experimental systems to investigate ER stress and UPR signaling in immune contexts:
In vitro models frequently utilize human cell lines such as CaCo-2 intestinal epithelial cells stimulated with gliadin peptides to study UPR involvement in inflammatory disease processes [98]. Primary immune cells isolated from blood or tissues can be treated with ER stress inducers to examine cell-type-specific UPR responses.
Ex vivo models include the mouse gut-ex-vivo system (GEVS), where intestinal tissue from gluten-free diet-fed mice is exposed to gliadin peptides, demonstrating prompt induction of ER stress markers in intestinal epithelial cells [98].
In vivo approaches incorporate genetic models such as myeloid-specific IRE1α knockout mice, which demonstrate protection from diet-induced obesity and insulin resistance through altered macrophage polarization [2] [19]. Disease models including diet-induced obesity or chemical-induced colitis help elucidate UPR contributions to pathophysiology.
Research Reagent Solutions
Table 3: Essential Reagents for UPR and Immune Function Research
| Reagent/Category | Specific Examples | Function/Application | Key Findings Enabled |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin (Tm), Thapsigargin (Tg) | experimentally induce ER stress; Tm inhibits N-linked glycosylation; Tg disrupts ER calcium homeostasis [2] [19] | established causal links between ER stress and inflammatory cytokine production in immune cells [2] |
| UPR Pathway Inhibitors | STF-083010 (IRE1α RNase inhibitor) | selectively inhibits IRE1α's RNase activity, blocking XBP1 splicing [2] [19] | demonstrated that IRE1α inhibition ameliorates insulin resistance and reduces pro-inflammatory macrophages in obesity [2] |
| Chemical Chaperones | 4-PBA, TUDCA | improve ER folding capacity and reduce ER stress [98] | prevented gliadin-induced pathophysiology in celiac disease models [98] |
| Genetic Models | Myeloid-specific IRE1α knockout mice | enables cell-type-specific investigation of UPR pathway function [2] | established that IRE1α in myeloid cells promotes pro-inflammatory M1 macrophage polarization [2] |
| Pathogen-Associated Molecular Patterns | LPS (bacterial), Zymosan (fungal) | immune cell stimulation in combination with ER stress [2] | revealed pathogen-specific UPR branch utilization (PERK for LPS; IRE1α for zymosan) in dendritic cells [2] |
Therapeutic Targeting of UPR in Immune-Mediated Diseases
The strategic modulation of UPR signaling represents a promising therapeutic approach for various immune-mediated diseases. Potential strategies include:
IRE1α-Targeted Therapeutics: Pharmacological inhibition of IRE1α's RNase activity with compounds like STF-083010 has shown efficacy in reducing adipose tissue inflammation and improving insulin sensitivity in murine models of obesity [2] [19]. This approach specifically targets the pro-inflammatory functions of IRE1α in macrophages without completely abrogating essential UPR signaling.
Chemical Chaperones: Small molecules such as 4-PBA and TUDCA can buffer ER stress by improving the ER's protein-folding capacity [98] [91]. In celiac disease models, chemical chaperones prevented gliadin-induced UPR activation, inflammation, and barrier dysfunction, suggesting potential clinical utility [98].
Natural Product Modulators: Various natural compounds demonstrate UPR-modulating activities through diverse mechanisms [91]. These include compounds that act as UPR inhibitors, activators, or that indirectly alleviate ER stress by reducing oxidative burden or improving protein folding capacity [91]. Their pleiotropic effects may offer advantages for modulating complex UPR-associated disease pathways.
Integrated UPR-Immunotherapy Approaches: In cancer, UPR activation creates an immunosuppressive tumor microenvironment that inhibits T cell function and promotes pro-tumorigenic immune responses [17] [99]. Combining UPR modulators with immune checkpoint inhibitors may therefore enhance anti-tumor immunity and overcome therapeutic resistance [17].
UPR Pathway Visualization
UPR Signaling in Immune Regulation
This diagram illustrates how various stressors activate the three principal UPR sensor pathways, leading to diverse immune cell outcomes. The IRE1α-XBP1s axis predominantly enhances cytokine production and cell survival, while the PERK-ATF4-CHOP pathway drives both cytokine expression and macrophage polarization. ATF6 signaling primarily supports cellular survival functions, and IRE1α activation can directly promote NLRP3 inflammasome assembly in specific contexts.
The UPR has evolved from being considered primarily a protein quality control system to recognition as a central regulator of immune cell function and inflammatory processes. The cell-type-specific and context-dependent functions of ER stress responses reveal complex regulatory networks that influence immune cell fate, activation, polarization, and effector functions [2]. Understanding these mechanisms at greater resolution will be crucial for developing targeted therapeutic interventions.
Future research directions should include elucidating the precise molecular mechanisms underlying the switch from adaptive to pro-apoptotic UPR signaling in specific immune cell subsets, exploring the crosstalk between UPR pathways and other inflammatory signaling networks such as NF-κB, and developing more targeted delivery systems for UPR modulators to specific immune cell populations [2] [98] [54]. As our understanding of these mechanisms deepens, so too will our ability to therapeutically harness the UPR-immune axis for treating a wide spectrum of inflammatory diseases.
Navigating UPR Experimental Complexities: Troubleshooting and Data Interpretation
Common Pitfalls in UPR Marker Detection and Ensuring Specificity
The Unfolded Protein Response (UPR) is a critical signaling network activated by endoplasmic reticulum (ER) stress, mediated by three principal ER-resident sensors: IRE1, PERK, and ATF6. Accurate detection of UPR activation is fundamental to understanding its role in cellular physiology and disease pathogenesis, from cancer to neurodegenerative disorders. However, the complexity of UPR signaling, characterized by bidirectional cell fate decisions, dynamic temporal regulation, and cross-talk between pathways, presents significant technical challenges for specific marker detection. Researchers must navigate these complexities to avoid misinterpretation that could compromise experimental validity and biological conclusions. This guide addresses common pitfalls and provides robust methodological frameworks to ensure specificity in UPR marker detection, enabling more reliable investigation of this crucial stress response pathway.
UPR Signaling Pathways: Key Markers and Molecular Events
The Three Arms of the Unfolded Protein Response
The UPR is orchestrated by three ER-transmembrane sensors that initiate distinct signaling cascades to restore proteostasis. Under normal conditions, these sensors are maintained in an inactive state through association with the chaperone BiP (GRP78). The accumulation of unfolded or misfolded proteins in the ER lumen triggers BiP dissociation, leading to sensor activation through distinct mechanisms [3] [45].
PERK-eIF2α-ATF4 Pathway: PERK (Protein kinase R-like ER kinase) is a type I transmembrane protein that responds to ER stress by dimerizing and autophosphorylating. Active PERK phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α), leading to global translational attenuation while selectively promoting translation of activating transcription factor 4 (ATF4) mRNA. ATF4 then regulates genes involved in amino acid metabolism, antioxidant response, and apoptosis. Under prolonged stress, ATF4 induces expression of the pro-apoptotic factor CHOP (C/EBP homologous protein) [3] [45].
IRE1-XBP1 Pathway: IRE1 (Inositol-requiring enzyme 1) possesses both kinase and endoribonuclease activities. Upon ER stress, IRE1 oligomerizes and autophosphorylates, activating its RNase domain. Active IRE1 catalyzes the unconventional splicing of XBP1 mRNA, removing a 26-nucleotide intron and causing a frameshift that produces the active transcription factor XBP1s (spliced XBP1). XBP1s upregulates genes involved in ER biogenesis, ER-associated degradation (ERAD), and lipid synthesis [100] [3] [81].
ATF6 Pathway: ATF6 (Activating transcription factor 6) is a type II transmembrane protein that translocates to the Golgi apparatus under ER stress. There, it undergoes proteolytic cleavage by site-1 and site-2 proteases (S1P and S2P), releasing its cytosolic domain (ATF6f). This fragment functions as a potent transcription factor that upregulates ER chaperones and components of the ERAD pathway [3] [45].
The following diagram illustrates the core signaling pathways and key detection markers for the UPR:
Key Detection Markers and Their Biological Significance
Table 1: Primary UPR Markers and Their Detection Significance
| Pathway | Marker | Detection Method | Biological Significance | Dynamic Range |
|---|---|---|---|---|
| PERK | p-eIF2α | Western blot, immunofluorescence | Early response, translational control | Rapid phosphorylation (min), dephosphorylation with prolonged stress |
| PERK | ATF4 | Western blot, qPCR | Integrated stress response mediator | Transient accumulation (1-8h) |
| PERK | CHOP | Western blot, qPCR | Pro-apoptotic commitment | Late phase (4-24h), indicates severe stress |
| IRE1 | XBP1 splicing | RT-PCR, qPCR | IRE1 enzymatic activity | Early to mid-phase (30min-12h) |
| IRE1 | XBP1s protein | Western blot, immunofluorescence | Active transcriptional regulator | Mid-phase (2-16h) |
| ATF6 | Cleaved ATF6 (ATF6f) | Western blot | ATF6 proteolytic activation | Early phase (1-8h) |
| General | BiP/GRP78 | Western blot, qPCR | ER chaperone induction, stress magnitude | Early sustained increase (4-48h) |
The temporal dynamics of UPR marker expression create distinct activation patterns that reflect both the duration and intensity of ER stress. The PERK-eIF2α axis typically responds within minutes to hours, while IRE1 and ATF6 activation occurs within 1-8 hours. CHOP expression generally indicates prolonged, unresolved stress (4-24 hours). These temporal characteristics are crucial for experimental design and interpretation, as sampling at inappropriate timepoints may miss pathway activation or capture only terminal apoptotic signaling rather than adaptive responses [3] [45].
Common Pitfalls in UPR Marker Detection
Technical Artifacts and Methodological Limitations
Antibody Specificity Issues: A predominant challenge in UPR research is the validation of antibody specificity. Many commercial antibodies demonstrate cross-reactivity with unrelated proteins or fail to distinguish between modified and unmodified forms. For example, phospho-specific antibodies for p-eIF2α may cross-react with other phosphorylated eIF2 family members, while ATF6 antibodies must distinguish between full-length (90kDa) and cleaved (50kDa) forms. Without proper validation, false positives can mislead pathway activation assessment [3].
Inadequate Control for Stress Induction: The use of inappropriate positive controls represents another significant pitfall. Common ER stress inducers like tunicamycin (N-linked glycosylation inhibitor) and thapsigargin (SERCA pump inhibitor) have pleiotropic effects beyond UPR activation. Tunicamycin, for instance, disrupts multiple glycosylation-dependent processes, while thapsigargin profoundly alters calcium signaling. These off-target effects can activate stress responses independent of canonical UPR pathways, complicating data interpretation [101] [3].
XBP1 Splicing Detection Artifacts: The gold standard for IRE1 pathway assessment—detection of XBP1 mRNA splicing—is susceptible to methodological artifacts. Conventional RT-PCR approaches may fail to resolve the slight size difference between unspliced (XBP1u) and spliced (XBP1s) products, leading to inaccurate quantification. Additionally, restriction enzyme-based assays (using PstI) can produce incomplete digestion, while qPCR approaches require carefully validated primer sets that distinguish between XBP1u and XBP1s without cross-amplification [100] [81].
Biological Complexity and Interpretation Challenges
Pathway-Specific versus Global UPR Activation: Researchers often overinterpret individual marker changes as representing global UPR activation. However, the three UPR branches can be differentially activated depending on stress modality, cell type, and physiological context. For example, lipid disequilibrium preferentially activates IRE1, while protein glycosylation defects may predominantly engage PERK signaling. This differential regulation means that monitoring only one pathway provides an incomplete picture of UPR status [101] [81].
Adaptive versus Pro-apoptotic Signaling: A critical interpretive challenge lies in distinguishing adaptive UPR signaling from terminal pro-apoptotic responses. Chronic ER stress induces a switch from protective to apoptotic signaling through mechanisms such as PERK-ATF4-CHOP activation and IRE1-TRAF2-ASK1-JNK pathway engagement. Without temporal analysis and multiple marker assessment, researchers may incorrectly categorize apoptotic signaling as adaptive UPR activation [3] [45].
Cell Type-Specific UPR Heterogeneity: Different cell types exhibit substantial variation in UPR component expression and activation thresholds. Immune cells, particularly dendritic cells and macrophages, demonstrate heightened IRE1-XBP1 signaling dependency, while professional secretory cells like pancreatic β-cells rely on all three branches. This heterogeneity means that detection strategies optimized for one cell type may perform poorly in others [2] [47].
Experimental Strategies for Ensuring Specificity
Multi-Parameter UPR Assessment Framework
Robust UPR analysis requires orthogonal approaches that collectively validate pathway activation. The following experimental workflow provides a comprehensive framework for specific UPR detection:
Integrated Workflow for Specific UPR Pathway Detection:
Protocol for Comprehensive XBP1 Splicing Analysis
Principle: Detection of IRE1-mediated XBP1 mRNA splicing provides the most specific measurement of IRE1 pathway activation. This protocol combines conventional RT-PCR with restriction digest validation for maximum specificity.
Reagents and Equipment:
- TRIzol or equivalent RNA isolation reagent
- DNase I (RNase-free)
- Reverse transcription system with random hexamers
- PCR reagents and thermocycler
- XBP1 primers: Forward 5'-AAACAGAGTAGCAGCTCAGACTGC-3', Reverse 5'-TCCTTCTGGGTAGACCTCTGGGAG-3'
- PstI restriction enzyme and buffer
- Agarose gel electrophoresis system
- Capillary electrophoresis system (optional, for higher resolution)
Procedure:
- RNA Isolation and DNase Treatment: Isolate high-quality RNA using TRIzol reagent. Treat with DNase I to eliminate genomic DNA contamination. Verify RNA integrity by agarose gel electrophoresis (sharp 18S and 28S ribosomal bands).
cDNA Synthesis: Perform reverse transcription using 500ng-1μg total RNA with random hexamers. Include a no-reverse-transcriptase (-RT) control for each sample to detect genomic DNA contamination.
PCR Amplification: Amplify XBP1 cDNA using the following conditions:
- 95°C for 3 min (initial denaturation)
- 35 cycles of: 95°C for 30s, 60°C for 30s, 72°C for 45s
- 72°C for 5 min (final extension)
Restriction Digest Analysis: Digest half of the PCR product with PstI restriction enzyme (2-4 hours, 37°C). XBP1u contains a PstI site that is lost upon splicing, allowing differentiation of spliced and unspliced products.
Product Resolution: Separate digested and undigested PCR products on a 3% agarose gel. XBP1u produces 289bp and 194bp fragments after PstI digestion, while XBP1s remains uncut (483bp). Alternatively, use capillary electrophoresis for superior resolution and quantification.
Interpretation and Troubleshooting:
- A predominance of the 483bp band (XBP1s) indicates IRE1 activation
- The -RT control should show no amplification
- Inefficient PstI digestion suggests enzyme activity issues or reaction optimization needs
- Faint or absent bands may indicate poor RNA quality or suboptimal PCR conditions
- For quantitative applications, combine with qPCR using spliced-specific primers [100] [81]
The Scientist's Toolkit: Essential Reagents and Controls
Table 2: Key Research Reagent Solutions for UPR Detection
| Category | Reagent/Control | Specific Function | Validation Requirements |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin (0.5-5μg/mL) | N-linked glycosylation inhibitor | Verify GRP78 induction and eIF2α phosphorylation |
| Thapsigargin (0.1-1μM) | SERCA pump inhibitor | Confirm ATF6 processing and XBP1 splicing | |
| Brefeldin A (1-10μg/mL) | ER-Golgi transport disruptor | Assess ATF6 activation | |
| Pathway Inhibitors | GSK2606414 (PERKi, 0.1-1μM) | PERK kinase inhibitor | Validate p-eIF2α reduction |
| 4μ8C (IRE1i, 10-100μM) | IRE1 RNase inhibitor | Confirm XBP1 splicing blockade | |
| Ceapins (ATF6i, 1-10μM) | ATF6 activation inhibitor | Verify blocked ATF6 cleavage | |
| Genetic Controls | siRNA/shRNA knockdown | Pathway component depletion | Confirm target protein reduction >70% |
| XBP1-/- cells | IRE1 pathway deficiency | Verify absent XBP1s protein | |
| ATF4-/- MEFs | PERK pathway deficiency | Confirm lack of CHOP induction | |
| Critical Antibodies | Anti-p-eIF2α (S51) | Phospho-specific detection | Verify loss of signal with λ-phosphatase treatment |
| Anti-ATF6 (N-terminal) | Cleaved ATF6 detection | Confirm 50kDa fragment specificity | |
| Anti-XBP1s (spliced) | Spliced XBP1 detection | Validate specificity with XBP1-/- cells | |
| Validation Tools | λ-phosphatase treatment | Phospho-antibody validation | Confirm signal loss with phosphatase |
| CRISPR-Cas9 KO cells | Antibody specificity control | Verify absent signal in KO background | |
| Recombinant protein | Positive control for Western | Confirm expected molecular weight |
Advanced Techniques and Emerging Approaches
Single-Cell UPR Analysis
Traditional bulk analysis masks cell-to-cell heterogeneity in UPR activation, potentially obscuring important biological insights. Single-cell RNA sequencing (scRNA-seq) enables resolution of UPR pathway activation at the individual cell level, revealing subpopulations with distinct stress response signatures. For targeted analysis, flow cytometry panels incorporating XBP1s-specific antibodies and phospho-eIF2α detection allow high-throughput assessment of UPR heterogeneity in complex cell populations [2] [47].
Spatial UPR Detection in Tissue Contexts
Understanding UPR activation in pathological contexts requires preservation of spatial information. Multiplex immunofluorescence approaches using tyramide signal amplification (TSA) enable simultaneous detection of multiple UPR markers (e.g., p-eIF2α, XBP1s, CHOP) within tissue architecture. RNAscope technology provides robust in situ hybridization for XBP1s mRNA with single-molecule sensitivity, localizing IRE1 activation to specific cell types in complex tissues [102].
Functional UPR Assessment Beyond Marker Detection
While molecular markers indicate pathway activation, functional assessments provide critical validation of UPR physiological impact:
- ER Proteostasis Capacity: Fluorescence-based protein secretion reporters (e.g., Gaussia luciferase) quantify ER functional capacity
- ERAD Activity: Degradation kinetics of established ERAD substrates (e.g., null Hong Kong α1-antitrypsin) monitor ERAD function
- Mitochondrial-ER Crosstalk: Calcium flux assays and APEX-based proximity labeling assess UPR-mediated organelle communication [17] [103]
Accurate detection of UPR activation requires meticulous experimental design, orthogonal validation approaches, and careful interpretation contextualized by biological understanding. The inherent complexity of UPR signaling—with its branch-specific dynamics, cell-type variations, and context-dependent outcomes—demands sophisticated methodological frameworks beyond single-marker analysis. By implementing the comprehensive strategies outlined in this guide, researchers can overcome common pitfalls and generate robust, reproducible data that advances our understanding of ER stress biology across diverse physiological and pathological contexts. As UPR-targeted therapies progress toward clinical application, particularly in cancer and neurodegenerative diseases, precise UPR biomarker detection becomes increasingly critical for both basic research and translational applications [17] [81] [45].
Optimizing Western Blot Analysis for Phospho-IRE1α (Ser724) and Other Low-Abundance Targets
The detection of phosphorylated IRE1α at Ser724 serves as a critical biomarker for monitoring the activation of the Inositol-Requiring Enzyme 1α (IRE1α) branch of the unfolded protein response (UPR). This technical guide provides comprehensive, optimized methodologies for resolving the challenges associated with Western blot analysis of this and other low-abundance targets. Within the broader context of endoplasmic reticulum (ER) stress research, precise detection of phospho-IRE1α (Ser724) is essential for understanding UPR signaling dynamics and its implications in diseases such as metabolic disorders and cancer. This whitepaper synthesizes current protocols to deliver robust, reproducible techniques for researchers and drug development professionals, focusing on sample preparation, protein separation, transfer efficiency, and immunodetection specific to the ~110 kDa IRE1α protein.
The endoplasmic reticulum (ER) is a crucial organelle responsible for the synthesis, folding, and post-translational modification of nearly one-third of the cellular proteome. The accumulation of unfolded or misfolded proteins within the ER lumen triggers ER stress, activating an adaptive signaling network known as the unfolded protein response (UPR). IRE1α is an evolutionarily conserved ER-transmembrane sensor and the most ancient branch of the UPR. It possesses dual serine/threonine protein kinase and endoribonuclease (RNase) activities in its cytoplasmic domain. Upon ER stress, IRE1α undergoes dimerization, oligomerization, and autophosphorylation, leading to the activation of its RNase domain. This activity catalyzes the unconventional splicing of XBP1 mRNA, producing a potent transcription factor (sXBP1) that drives the expression of chaperones and components of ER-associated degradation (ERAD) to restore proteostasis.
Phosphorylation at Ser724, located within the kinase activation loop of IRE1α, is a pivotal regulatory event. A recent study utilizing an Ern1S724A/S724A knock-in mouse model demonstrated that ablation of Ser724 phosphorylation markedly reduces IRE1α autophosphorylation and blunts its RNase activity, as measured by Xbp1 mRNA splicing. This mutation also exacerbated hepatic steatosis in mice under ER stress, highlighting the critical role of this phosphorylation site in dynamically controlling IRE1α's enzymatic output and its physiological function in metabolic regulation [104]. Consequently, accurate detection of phospho-IRE1α (Ser724) is indispensable for UPR research, yet it presents significant technical challenges due to its transient nature, low abundance, and the necessity of preserving post-translational modifications during sample preparation.
Technical Challenges and Optimization Principles
Detecting low-abundance proteins like phospho-IRE1α via Western blot requires enhanced sensitivity and optimized conditions to overcome inherent obstacles. Standard protocols often yield insufficient signal for targets present in minimal quantities. The fundamental principles for optimization encompass every stage of the workflow:
- Maximizing Signal-to-Noise Ratio: The core challenge is to amplify the specific signal from the target protein while minimizing background interference. This involves using a higher concentration of primary antibody, optimizing blocking conditions, and selecting high-sensitivity detection substrates [105] [106].
- Preserving Post-Translational Modifications: Phospho-epitopes are labile. Sample preparation must include phosphatase inhibitors to prevent enzymatic dephosphorylation during and after cell lysis [105]. Additionally, selecting an appropriate blocking agent (e.g., BSA over milk) is crucial to avoid background from phospho-proteins present in milk [107].
- Ensuring Protein Integrity and Transfer Efficiency: Low-abundance proteins are susceptible to degradation and inefficient transfer. Using protease inhibitors, selecting neutral-pH gel chemistries that preserve protein integrity, and employing optimized transfer methods for high molecular weight proteins (~110 kDa for IRE1α) are essential steps [105] [106].
The following diagram illustrates the core optimization strategy for low-abundance targets, connecting the central challenge to the main goals and the specific techniques used to achieve them.
Optimized Experimental Protocols
Sample Preparation for Phospho-Protein Analysis
Effective sample preparation is the foundation for successful detection of phospho-IRE1α, designed to preserve the phosphorylation state and maximize protein yield [105].
Detailed Protocol:
- Cell Stimulation and Lysis:
- Induce ER stress in cultured cells using chemical inducers such as Tunicamycin (Tm, 1-5 µg/mL) or Thapsigargin (Tg, 1-5 µM) for 3-6 hours [104]. For phospho-IRE1α, specific positive controls can be generated by treating Min6 cells with high glucose (e.g., 20 nM for 3 hours) or HeLa cells with Dithiothreitol (DTT, 10 mM for 1 hour) [107].
- Aspirate media and wash cells twice with ice-cold PBS.
- Lyse cells directly in plates on ice using RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with:
- 5% (v/v) protease inhibitor cocktail
- 1-2% (v/v) phosphatase inhibitor cocktail [105]
- For tissues, manually macerate and homogenize in lysis buffer at a 1:10 (w/v) ratio using a Dounce homogenizer or electric homogenizer [108].
Clarification and Protein Quantification:
- Scrape lysates, transfer to microcentrifuge tubes, and incubate on ice for 15 minutes.
- Sonicate the lysates (e.g., 3s pulse, 10s interval, 5-15 times at 40 kW) to disrupt nucleic acids and release nuclear proteins, then centrifuge at 14,000–17,000 x g for 15 minutes at 4°C [105].
- Transfer the supernatant to a fresh tube. Determine protein concentration using a BCA or Bradford assay, ensuring a standard curve with an R-squared value ≥ 0.99 for accuracy [108].
Sample Denaturation:
- Mix lysate with 5× Laemmli sample buffer to avoid excessive dilution [105].
- For IRE1α and other transmembrane proteins, avoid boiling to prevent aggregation. Instead, incubate samples at 70°C for 10-20 minutes, or at 37°C for 30 minutes [105].
- Use freshly prepared lysates immediately or store at -80°C. Avoid freeze-thaw cycles for phospho-proteins.
Table 1: Recommended Inhibitor Cocktails for Phospho-IRE1α Analysis
| Inhibitor Type | Recommended Concentration | Purpose | Example Product |
|---|---|---|---|
| Protease Inhibitor Cocktail | 5% (v/v) in lysis buffer | Prevents degradation of IRE1α protein by cellular proteases | Broad-spectrum cocktails (e.g., EDTA-free) |
| Phosphatase Inhibitor Cocktail | 1-2% (v/v) in lysis buffer | Preserves Ser724 phosphorylation by inhibiting cellular phosphatases | Sodium fluoride, beta-glycerophosphate, sodium orthovanadate |
Gel Electrophoresis and Protein Separation
Optimal separation is critical for resolving the ~110 kDa IRE1α protein from confounding bands.
Detailed Protocol:
- Gel Selection: Use 8% or 4-12% gradient Bis-Tris gels for optimal resolution of IRE1α. Bis-Tris gels with a neutral pH preserve protein integrity and minimize modifications, leading to sharper bands and superior transfer efficiency compared to traditional Tris-glycine gels [107] [106].
- Sample Loading: Load 50-100 µg of total protein per lane for low-abundance targets. Use a 1.5 mm thick comb to increase loading capacity. Include a pre-stained protein ladder and a confirmed positive control lysate in each run [105] [107].
- Electrophoresis Conditions:
- Prepare MES or MOPS running buffer. MES is preferred for optimal resolution in the 3.5-160 kDa range.
- Load samples and run gels at a constant voltage. Begin with 80 V for 4-5 minutes to allow proteins to enter the stack uniformly, then increase to 180 V for approximately 50 minutes, or until the dye front reaches the bottom of the gel [108].
Protein Transfer and Membrane Blocking
Efficient transfer of high molecular weight proteins like IRE1α requires specific optimization to ensure complete movement from the gel to the membrane.
Detailed Protocol:
- Membrane Selection: Use PVDF membranes for their high protein-binding capacity, which is advantageous for low-abundance proteins. Pre-wet PVDF membranes in 100% methanol for 1 minute, then equilibrate in transfer buffer [105].
- Transfer Method: For IRE1α (~110 kDa), employ a wet transfer system at 4°C.
- To enhance transfer efficiency, the transfer buffer can be supplemented with SDS to a final concentration of ≤ 0.1% [107].
- Transfer can be extended (e.g., 90 minutes to 2 hours) to ensure complete movement of the high molecular weight protein.
- As an alternative, dry electroblotting systems with ready-to-use stacks offer convenience and high transfer consistency [106].
- Transfer Success Check: After transfer, stain the membrane with Ponceau S for 1-10 minutes to visualize protein bands and confirm uniform transfer, particularly in the 90-130 kDa region. Wash with distilled water or TBST to destain before blocking [105] [107].
- Blocking: To avoid high background common with phospho-specific antibodies, block the membrane for 1 hour at room temperature with 5% Bovine Serum Albumin (BSA) in TBST. Do not use milk-based blockers, as they contain phospho-proteins that can cause elevated background [107].
Immunodetection and Signal Amplification
This stage is crucial for generating a specific, strong signal from the low-abundance phospho-protein.
Detailed Protocol:
- Primary Antibody Incubation:
- Use a validated phospho-IRE1α (Ser724) antibody (e.g., Rabbit Polyclonal, Product # PA5-85738).
- Dilute the primary antibody in 1% BSA in TBST at a higher concentration than standard. Check the datasheet and reduce the dilution ratio; a starting point of 1:500 to 1:1000 is often appropriate [105] [109].
- Incubate the membrane with the primary antibody overnight at 4°C with gentle shaking [105].
- Washing: Wash the membrane three times for 5 minutes each with 1x TBST at room temperature to remove unbound primary antibody.
- Secondary Antibody Incubation:
- Use an HRP-conjugated secondary antibody specific to the host species of the primary antibody (e.g., Goat anti-Rabbit IgG).
- Dilute the antibody in 1% BSA/TBST at a higher concentration and incubate for 1 hour at room temperature with shaking.
- Ensure no sodium azide is present in any solution, as it inhibits HRP activity [105].
- Detection:
- For maximum sensitivity, use a high-sensitivity chemiluminescent substrate (e.g., SuperSignal West Atto Ultimate Sensitivity Substrate). These substrates can provide over 3x more sensitivity than conventional ECL and enable detection down to the attogram level [106].
- Image the blot using a digital imaging system with a CCD camera, which offers a wider dynamic range and better quantitation than traditional film.
Table 2: Key Research Reagent Solutions for Phospho-IRE1α Western Blot
| Reagent / Material | Function / Rationale | Optimized Recommendation |
|---|---|---|
| Phosphatase Inhibitor Cocktail | Preserves labile phosphorylation at Ser724 during lysis | Add 1-2% (v/v) to lysis buffer |
| Protease Inhibitor Cocktail | Prevents degradation of the IRE1α protein target | Add 5% (v/v) to lysis buffer |
| Bis-Tris or Tris-Acetate Gels | Neutral pH gels for superior protein integrity and resolution of high MW proteins | 8% or 4-12% gradient gels |
| PVDF Membrane | High protein-binding capacity for low-abundance targets | Pre-wet in methanol before use |
| Phospho-IRE1α (Ser724) Antibody | Primary antibody for specific detection of the activated protein | Use at 1:500-1:1000 dilution in 1% BSA/TBST |
| BSA Blocking Buffer | Reduces background for phospho-specific antibodies; avoids interference from milk phospho-proteins | 5% BSA in TBST, block for 1 hour |
| HRP-conjugated Secondary Antibody | Enables high-sensitivity chemiluminescent detection | Use at a higher concentration in 1% BSA/TBST |
| High-Sensitivity Chemiluminescent Substrate | Signal detection reagent for very low-abundance targets | e.g., SuperSignal West Atto |
Verification and Data Analysis
Accurate interpretation of phospho-IRE1α blots requires careful normalization and control procedures.
- Total Protein Normalization: To ensure that changes in phospho-IRE1α signal reflect true activation and not merely variations in total IRE1α protein or loading errors, the membrane must be re-probed for total IRE1α. After detecting phospho-IRE1α, strip the membrane and re-probe it with an antibody against total IRE1α (e.g., NB100-2324) [107]. The ratio of phospho-signal to total protein signal provides the most accurate measure of activation.
- Loading Controls: Use a mid-to-high molecular weight loading control, such as Tubulin, to verify equal protein loading across all lanes [107].
- Troubleshooting: If high background is observed, include a secondary antibody-only control (no primary antibody) to confirm the source is not the secondary antibody itself [107].
The following workflow provides a consolidated summary of the optimized protocol from sample to image.
Concluding Remarks
Within the framework of UPR and ER stress research, the ability to reliably detect phospho-IRE1α (Ser724) is fundamental to elucidating the molecular mechanisms of diseases ranging from metabolic syndrome to neurodegeneration and cancer. The optimized protocols detailed in this guide, from stringent sample preparation with specific inhibitors to the use of high-sensitivity detection systems, provide a robust methodological foundation. Adherence to these guidelines, including rigorous normalization and validation, will enable researchers and drug developers to generate reproducible, quantitative data on IRE1α activation, thereby advancing our understanding of ER stress biology and contributing to the development of novel therapeutic strategies.
The Unfolded Protein Response (UPR) is a critical cellular signaling network activated by Endoplasmic Reticulum (ER) stress, a state triggered by the accumulation of unfolded or misfolded proteins in the ER lumen [110] [5]. Research into the UPR has expanded dramatically due to its established role in diverse human diseases, including neurodegenerative disorders, diabetes, cancer, and inflammatory conditions [2] [110]. However, the complexity of the UPR, comprising three dynamically regulated branches (PERK, IRE1α, and ATF6), presents significant challenges for experimental accuracy and reproducibility [110] [111]. This technical guide underscores the indispensable role of stressed cell lysates as positive controls, providing a foundational strategy to validate experimental systems, ensure accurate interpretation of UPR activation, and ultimately generate reliable, publication-quality data for researchers and drug development professionals.
The absence of appropriate positive controls is a critical flaw that can compromise data integrity. Without a validated benchmark, it is impossible to distinguish between negative results caused by experimental failures and genuine biological phenomena. Stressed cell lysates, generated by treating cells with well-characterized ER stress inducers, provide an essential internal control for techniques ranging from Western blotting and RT-qPCR to reporter assays [111]. This guide provides a detailed methodology for the preparation, validation, and application of these lysates, framing them within the broader context of robust UPR research practices.
UPR Signaling Pathways: A Primer for Experimental Design
A thorough understanding of UPR signaling is a prerequisite for designing experiments and selecting appropriate readouts for positive controls. The UPR is orchestrated by three ER-transmembrane sensors: PERK, IRE1α, and ATF6. Under normal conditions, the ER chaperone BiP (GRP78) binds to these sensors, maintaining them in an inactive state. The accumulation of unfolded proteins recruits BiP away from the sensors, triggering their activation [110] [5].
Figure 1: The Three Branches of the Unfolded Protein Response. This diagram illustrates the core signaling pathways of the UPR, initiated by ER stress. The activation of PERK, IRE1α, and ATF6 leads to distinct transcriptional and translational outputs that determine cell fate, balancing adaptive survival and pro-apoptotic signals. Understanding these pathways is essential for selecting validation markers for stressed cell lysates.
Generating Stressed Cell Lysates: A Standardized Protocol
The following protocol details the generation of stressed cell lysates suitable for use as positive controls in a wide array of molecular biology techniques.
Materials and Reagents
Table 1: Common Pharmacological ER Stress Inducers and Their Mechanisms of Action
| Inducer | Mechanism of Action | Typical Working Concentration | Key UPR Pathways Activated |
|---|---|---|---|
| Tunicamycin | Inhibits N-linked glycosylation, causing accumulation of unglycosylated/unfolded proteins [111] | 2.5 - 5 µg/mL for 5 hours [111] | PERK, IRE1α, ATF6 |
| Thapsigargin | Inhibits SERCA pump, depleting ER calcium stores and disrupting calcium-dependent chaperones [111] | 0.1 - 1 µM for 5 hours [111] | PERK, IRE1α, ATF6 |
| Dithiothreitol (DTT) | A reducing agent that disrupts disulfide bond formation in the ER [112] [111] | 1 - 5 mM for 30 minutes to 2 hours [111] | IRE1α, PERK |
| Brefeldin A | Disrupts ER-to-Golgi transport, causing proteins to accumulate in the ER [111] | 5 - 10 µg/mL for 5 hours [111] | IRE1α, ATF6 |
Step-by-Step Experimental Procedure
Cell Culture and Seeding: Use a standard mammalian cell line relevant to your research. Mouse Embryonic Fibroblasts (MEFs) are well-characterized and many UPR component knockout lines are available [111]. For professional secretory cells, consider β-cell lines (e.g., MIN6, INS-1) or plasma cell-derived lines (e.g., J558) [111].
- Seed cells at an appropriate density (e.g., 70-80% confluency) in tissue culture plates or dishes to ensure they are in an active growth phase during treatment.
ER Stress Induction:
- Prepare fresh stock solutions of the chosen inducer(s) in a suitable solvent (e.g., DMSO for tunicamycin and thapsigargin; water for DTT).
- Replace the cell culture medium with fresh medium containing the predetermined concentration of the ER stress inducer. Include a vehicle control (e.g., 0.1% DMSO) for untreated samples.
- Incubate cells for the optimized time period at 37°C and 5% CO₂. The duration can range from 30 minutes for rapid markers like eIF2α phosphorylation to several hours for markers like CHOP and XBP1 splicing [111].
Cell Lysis and Lysate Preparation:
- Post-incubation, place the culture dish on ice and quickly aspirate the medium.
- Wash cells twice with ice-cold Phosphate-Buffered Saline (PBS).
- Add an appropriate lysis buffer (e.g., RIPA buffer) supplemented with protease and phosphatase inhibitors directly to the cells.
- Scrape the cells and transfer the lysate to a microcentrifuge tube. Incubate on ice for 15-30 minutes with occasional vortexing.
- Clarify the lysate by centrifugation at >12,000 × g for 15 minutes at 4°C.
- Carefully transfer the supernatant (the total cell lysate) to a new pre-chilled tube.
Quality Control and Storage:
- Determine the protein concentration of each lysate using a standard assay (e.g., BCA or Bradford).
- Aliquot the lysates to avoid repeated freeze-thaw cycles and store at -80°C for long-term use.
Validating Stressed Cell Lysates: Key Markers and Methods
Once prepared, the stressed cell lysates must be rigorously validated to confirm successful UPR induction. The table below summarizes the key molecular markers across the three UPR branches that should be assessed.
Table 2: Essential UPR Markers for Validating Stressed Cell Lysates
| UPR Branch | Key Marker | Detection Method | Expected Result in Positive Control | Function/Biological Significance |
|---|---|---|---|---|
| PERK | p-eIF2α (Ser51) | Western Blot | Increased phosphorylation [112] [111] | Attenuates global translation [110] [111] |
| ATF4 | Western Blot / RT-qPCR | Increased protein & mRNA levels [112] | Transcription factor for stress response [110] | |
| CHOP (DDIT3) | Western Blot / RT-qPCR | Increased protein & mRNA levels [112] [113] | Pro-apoptotic transcription factor [110] [113] | |
| IRE1α | XBP1 mRNA Splicing | RT-PCR / Gel Electrophoresis | Shift to spliced variant (XBP1s) [112] | Active transcription factor for ER biogenesis [110] |
| XBP1s Protein | Western Blot | Increased nuclear protein [2] | Drives chaperone and lipid synthesis gene expression [110] | |
| ATF6 | Cleaved ATF6 (ATF6-p50) | Western Blot | Appearance of ~50 kDa fragment [110] | Active nuclear transcription factor [110] |
| General/Multiple | BiP (GRP78) | Western Blot / RT-qPCR | Increased protein & mRNA levels [112] [111] | Master ER chaperone; central UPR regulator [110] [111] |
Workflow for Comprehensive Validation
A multi-faceted approach is recommended for thorough validation, as depicted in the workflow below.
Figure 2: A Multi-Method Workflow for Validating Stressed Cell Lysates. This workflow illustrates the complementary techniques required to confirm UPR activation across different branches, from protein phosphorylation and expression to mRNA splicing events.
The Scientist's Toolkit: Essential Reagents for UPR Research
Table 3: Research Reagent Solutions for UPR Studies
| Reagent / Material | Function / Application | Example Use-Case |
|---|---|---|
| Tunicamycin | Inhibits N-linked glycosylation; induces ER stress via accumulation of unglycosylated proteins [111] | Classic inducer for robust, full-branch UPR activation; ideal for generating positive controls [111]. |
| Thapsigargin | SERCA pump inhibitor; induces ER stress by disrupting ER calcium homeostasis [111] | Rapid inducer of UPR; useful for studying calcium's role in ER stress and the PERK pathway [111]. |
| Anti-BiP/GRP78 Antibody | Detects levels of the master ER chaperone; key marker for UPR activation [111] [113] | Western blot validation of stressed lysates; immunohistochemistry to localize ER stress in tissues. |
| Anti-p-eIF2α (Ser51) Antibody | Detects phosphorylation of eIF2α; specific marker for PERK pathway activation [111] | Essential for confirming the earliest events in the PERK branch in Western blots of stressed lysates. |
| Anti-CHOP Antibody | Detects the pro-apoptotic transcription factor; marker for severe/prolonged ER stress [111] [113] | Distinguishing adaptive UPR from terminal UPR in validation assays; apoptosis linkage studies. |
| XBP1 Splicing Assay Kits | Detect the unconventional splicing of XBP1 mRNA; gold-standard marker for IRE1α activation [111] | Validating IRE1α branch activity in stressed lysates via RT-PCR and gel electrophoresis. |
| IRE1α RNase Inhibitors (e.g., STF-083010) | Selectively inhibits the RNase activity of IRE1α [2] | Tool for dissecting IRE1α-specific contributions to UPR and cell fate in mechanistic studies. |
The integration of well-characterized stressed cell lysates as positive controls is not a mere technical suggestion but a fundamental requirement for rigorous UPR research. These controls serve as an internal benchmark, enabling researchers to verify that their experimental systems are capable of detecting UPR activation and to accurately interpret their results, particularly negative ones. The protocols and validation strategies outlined in this guide provide a clear roadmap for generating and utilizing these critical reagents. By adopting these practices, the scientific community can enhance the reliability, reproducibility, and translational impact of its work in understanding the UPR's role in health and disease.
The unfolded protein response (UPR) represents a critical cellular signaling network that determines cell fate following endoplasmic reticulum (ER) stress. This sophisticated quality control mechanism demonstrates remarkable duality—initiating adaptive, pro-survival programs under transient stress conditions while activating terminal, pro-apoptotic pathways when ER homeostasis cannot be restored. The transition from adaptive to terminal UPR involves complex molecular switches influenced by stress intensity, duration, and cellular context. This technical review delineates the key mechanistic and quantitative distinctions between these opposing UPR phases, providing researchers with advanced experimental frameworks for their discrimination. Through comprehensive analysis of UPR signaling dynamics, biomarker profiles, and functional outcomes, we establish a foundational guide for investigating UPR in disease pathogenesis and therapeutic development.
The endoplasmic reticulum serves as the primary intracellular organelle responsible for protein folding, lipid synthesis, and calcium storage [91]. Its functional integrity is maintained through stringent quality control systems, particularly the unfolded protein response (UPR), which activates when the protein folding capacity is overwhelmed by unfolded or misfolded proteins [91] [3]. The UPR is orchestrated by three ER-resident transmembrane sensors: PERK (PKR-like ER kinase), IRE1α (inositol-requiring enzyme 1 alpha), and ATF6 (activating transcription factor 6) [91] [110]. Under normal conditions, these sensors remain inactive through association with the chaperone protein BiP/GRP78 [3] [114].
During ER stress, BiP dissociates to bind unfolded proteins, activating the UPR sensors and initiating a complex decision-making process that determines cellular survival [114]. The UPR's dual role as both protector and executioner represents one of its most fascinating characteristics—initially functioning to restore proteostasis through adaptive mechanisms, but triggering apoptosis if stress persists [91] [114]. This balance is crucial in various human diseases, including neurodegenerative disorders [114], cancer [17] [88], cardiovascular conditions [115], and metabolic syndromes [91]. The following sections provide a detailed examination of the molecular mechanisms distinguishing adaptive from terminal UPR, along with experimental approaches for their investigation.
Molecular Mechanisms and Signaling Switches
The Three UPR Sensor Pathways and Their Dual Outputs
Each UPR sensor activates distinct signaling branches with context-dependent outcomes:
PERK-eIF2α Pathway: PERK activation leads to phosphorylation of eukaryotic initiation factor 2α (eIF2α), resulting in global translational attenuation to reduce protein loading [91] [110]. This simultaneously enables selective translation of activating transcription factor 4 (ATF4), which coordinates expression of genes involved in amino acid metabolism, antioxidant responses, and ER stress resolution [91] [115]. Under persistent stress, ATF4 induces the pro-apoptotic transcription factor CHOP (C/EBP homologous protein), which regulates genes promoting oxidative stress and apoptosis [91] [110] [115].
IRE1α-XBP1 Pathway: IRE1α possesses dual kinase and endoribonuclease activities. Its activation splices a 26-nucleotide intron from XBP1 mRNA, generating the potent transcription factor XBP1s [91] [110]. XBP1s enhances ER folding capacity, lipid biosynthesis, and ER-associated degradation (ERAD) [91] [88]. However, under irreversible stress, IRE1α switches from XBP1 splicing to regulated IRE1-dependent decay (RIDD), degrading specific mRNAs and microRNAs, and activates pro-apoptotic JNK signaling through TRAF2-ASK1 complex formation [3] [88].
ATF6 Pathway: ER stress triggers ATF6 translocation to the Golgi apparatus, where it undergoes proteolytic cleavage by S1P and S2P proteases [110]. The released ATF6(N) cytosolic fragment functions as a transcription factor that upregulates ER chaperones (including BiP) and components of the ERAD machinery [91] [110]. While primarily adaptive, chronic ATF6 signaling may collaborate with other UPR branches to promote apoptosis under severe stress conditions [114].
Key Molecular Switches Determining UPR Outcome
The transition from adaptive to terminal UPR involves several critical molecular switches:
Table 1: Molecular Switches Governing Adaptive vs. Terminal UPR Transitions
| Molecular Component | Adaptive UPR Role | Terminal UPR Switch |
|---|---|---|
| PERK Signaling | Transient eIF2α phosphorylation and ATF4-mediated antioxidant response | Sustained PERK activation and CHOP-mediated apoptotic programming |
| IRE1α Activity | XBP1 splicing enhancing ER biogenesis and folding capacity | RIDD activation and TRAF2-ASK1-JNK pro-apoptotic signaling |
| CHOP Expression | Minimal or transient expression | Sustained upregulation downregulating BCL-2 and increasing oxidative stress |
| eIF2α Phosphorylation | Temporary translation attenuation to reduce ER load | Persistent phosphorylation with GADD34-mediated failed recovery |
| IRE1α Oligomerization | Transient oligomerization enabling XBP1 splicing | Hyper-oligomerization leading to RIDD dominance |
The integration of stress intensity and duration fundamentally determines UPR outcomes. Mild or transient stress typically engages adaptive mechanisms, while severe or prolonged stress triggers apoptotic signaling [114]. The CHOP-GADD34 regulatory loop exemplifies this switch: initially, GADD34 facilitates eIF2α dephosphorylation to restore translation, but chronic stress creates a feed-forward loop where CHOP maintains GADD34 expression, leading to excessive protein synthesis that overwhelms the stressed ER [3] [115].
The diagram below illustrates the core signaling pathways and molecular switches that differentiate adaptive from terminal UPR:
Quantitative Biomarkers and Differentiation Criteria
Differentiating adaptive from terminal UPR requires monitoring specific biomarkers with attention to their magnitude, timing, and combination. The table below summarizes key quantitative parameters for this discrimination:
Table 2: Quantitative Biomarkers for Differentiating Adaptive vs. Terminal UPR
| Parameter | Adaptive UPR | Terminal UPR | Measurement Methods |
|---|---|---|---|
| PERK Autophosphorylation | Transient (peaks 2-6h) | Sustained (>8-12h) | Phos-tag gel/Western [116] |
| XBP1 Splicing Ratio | Moderate increase (3-5 fold) | Initially high then declines | RT-PCR, Electrophoresis [56] |
| CHOP mRNA Level | Minimal increase (<5 fold) | Marked increase (>10 fold) | qPCR, RNA-seq [114] |
| ATF4 Protein | Transient (peaks 4-6h) | Sustained or biphasic | Western blot [56] |
| BiP/GRP78 Level | Gradual increase | Sharply elevated then may decline | Western blot, Immunofluorescence [114] |
| Caspase-12/-3 Activation | Absent or minimal | Significantly increased | Cleavage assays, Activity kits |
| JNK Phosphorylation | Low or transient | Sustained activation | Phospho-specific Western |
| Overall Protein Synthesis | Temporarily reduced (30-50%) | Persistently suppressed or erratic recovery | 35S-methionine incorporation [56] |
Temporal Dynamics as Discriminatory Factors
The timing and persistence of UPR activation serve as critical differentiators between adaptive and terminal phases. Adaptive UPR typically demonstrates transient sensor activation with rapid resolution following stress removal, while terminal UPR shows prolonged activation even after stress mitigation [114]. For instance, ATF4 protein levels typically peak at 4-6 hours post-stress induction in adaptive responses but remain elevated or display biphasic expression in terminal UPR [56]. Similarly, PERK autophosphorylation is transient during adaptation but sustained during apoptosis commitment [116].
The integration of multiple biomarkers provides more reliable differentiation than single-parameter assessments. For example, combined elevation of CHOP with JNK phosphorylation strongly indicates terminal UPR commitment, whereas isolated XBP1 splicing suggests adaptive signaling [3] [114]. Advanced analytical approaches, including multi-omics integration of transcriptomic, proteomic, and translatomic data, enable comprehensive assessment of UPR status across these parameters [56].
Experimental Approaches and Methodologies
Direct Monitoring of UPR Sensor Activation
Accurate differentiation of UPR phases requires direct assessment of sensor activation status rather than relying solely on downstream markers:
Phos-tag Gel Electrophoresis: This technique provides superior resolution for detecting shifts in protein mobility caused by phosphorylation of UPR sensors [116]. The methodology involves:
- Gel Preparation: Incorporate 25-50 μM Phos-tag acrylamide and 50-100 μM MnCl₂ into standard SDS-PAGE resolving gels [116].
- Sample Preparation: Lyse cells in Tris-based buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA) with protease and phosphatase inhibitors [116].
- Electrophoresis: Run at 100V for 3 hours for IRE1α or at 5mA for 9.5 hours for PERK detection [116].
- Transfer and Detection: Incubate gels in transfer buffer with 1 mM EDTA before standard Western blotting with anti-IRE1α or anti-PERK antibodies [116].
This approach enables direct visualization of IRE1α and PERK phosphorylation status, providing sensitive detection of even mild ER stress under physiological conditions [116].
XBP1 Splicing Assay: Monitor IRE1α activity through RT-PCR analysis of XBP1 splicing using specific primers flanking the splice site, followed by electrophoresis separation of spliced versus unspliced products [56]. The unconventional splicing of XBP1 mRNA serves as a specific indicator of IRE1α endoribonuclease activity.
Live-Cell Imaging and Unfolded Protein Monitoring
Recent advances in reporter systems enable real-time monitoring of UPR status in living cells:
Fluorescent UPR Reporters: Engineered PERK-based constructs that form fluorescence puncta upon unfolded protein binding allow direct visualization of unfolded protein accumulation in the ER lumen [117]. These systems demonstrate that persistent unfolded protein accumulation despite UPR attenuation predicts cellular apoptosis commitment [117].
Multiparameter Live-Cell Assays: Combine fluorescent reporters for UPR activation (e.g., XBP1-splicing reporters) with apoptosis markers (e.g., caspase activation sensors) to dynamically track the transition from adaptation to termination in individual cells.
Integrated Multi-Omics Approaches
Comprehensive UPR assessment benefits from integrated omics methodologies:
Ribosome Profiling: This technique enables genome-wide assessment of translation efficiency, particularly valuable for monitoring PERK-mediated translational control during ER stress [56]. It reveals selective mRNA translation despite global attenuation, identifying key mediators of UPR fate decisions.
Parallel Transcriptomics and Proteomics: Combined RNA sequencing and mass spectrometry analysis captures both rapid transcriptional responses and slower protein-level adaptations, providing systems-level understanding of UPR progression [56].
The experimental workflow for comprehensive UPR analysis integrates these approaches as illustrated below:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for UPR Pathway Investigation
| Reagent Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin (2.5 μg/ml), Thapsigargin (200 nM), Dithiothreitol (DTT) | Experimental ER stress induction [56] | N-glycosylation inhibition, SERCA pump inhibition, redox disruption |
| UPR Inhibitors | PERK inhibitor (e.g., GSK2606414), IRE1α RNase inhibitors (e.g., MKC-8866, B-I09), ATF6 inhibitors (e.g., Ceapins) | Branch-specific UPR perturbation [17] [88] | Pathway-specific functional dissection |
| Activation Detection | Anti-p-eIF2α, Anti-CHOP, Anti-XBP1s, Anti-BiP/GRP78 antibodies | UPR activation monitoring [114] | Downstream pathway output assessment |
| Sensor Phosphorylation Detection | Phos-tag acrylamide reagents, Anti-IRE1α, Anti-PERK antibodies | Direct sensor activation measurement [116] | Early UPR activation detection |
| Apoptosis Markers | Anti-cleaved Caspase-3, Caspase activity assays, Annexin V staining | Terminal UPR commitment confirmation [114] | Apoptotic pathway engagement |
| Live-Cell Reporters | PERK(LD)-EGFP-HOTag3, XBP1-splicing reporters [117] | Real-time UPR monitoring | Unfolded protein accumulation dynamics |
Pathophysiological Context and Therapeutic Implications
Disease-Specific UPR Transitions
The balance between adaptive and terminal UPR significantly influences disease progression across multiple pathological contexts:
Neurodegenerative Diseases: In Alzheimer's disease, initial Aβ and tau deposition induces mild ER stress and adaptive UPR activation [114]. As pathology advances, irreversible ER stress with maladaptive UPR promotes synaptic dysfunction and neuronal apoptosis [114]. Similar patterns occur in Parkinson's disease, where chronic UPR activation contributes to dopaminergic neuron loss.
Cancer Oncology: Tumor cells exploit adaptive UPR to survive microenvironmental stress (hypoxia, nutrient deprivation) [17] [88]. However, therapeutic interventions can push cancer cells beyond adaptive capacity into terminal UPR, providing opportunities for UPR-targeted therapies [17] [88]. PERK and IRE1α inhibitors are under investigation to disrupt pro-survival UPR signaling in tumors [17].
Cardiovascular Disorders: In cardiomyopathies, initial UPR activation serves adaptive functions, but sustained ER stress promotes maladaptive remodeling, cardiomyocyte apoptosis, and heart failure progression [115]. The transition involves CHOP-mediated apoptosis and disrupted calcium homeostasis.
Metabolic Diseases: In type 2 diabetes, chronic nutrient excess induces ER stress in pancreatic β-cells, with eventual transition from adaptive to terminal UPR contributing to β-cell dysfunction and apoptosis [91].
Therapeutic Targeting Strategies
Understanding the adaptive-terminal UPR transition enables several therapeutic approaches:
- UPR Hyperactivation: Push stressed cells beyond adaptive capacity into apoptosis (e.g., in cancer therapy) [17]
- UPR Inhibition: Protect vulnerable cells from terminal UPR commitment (e.g., in neurodegenerative disorders) [114]
- UPR Modulation: Fine-tune UPR signaling to maintain adaptive functions while preventing apoptosis [88]
- Combination Therapies: Co-target UPR pathways with conventional treatments to overcome resistance mechanisms [88]
The discrimination between adaptive and terminal UPR represents a critical research challenge with significant therapeutic implications. This differentiation requires multifactorial assessment of UPR sensor activation status, downstream signaling amplitude and duration, and integration of multiple biomarker readouts. The experimental frameworks outlined herein provide robust methodologies for investigating this cell fate decision across physiological and pathological contexts. Future research advances will require increasingly sophisticated single-cell analysis techniques to decode the heterogeneity of UPR responses within cell populations, ultimately enabling more precise therapeutic targeting of this fundamental cellular stress response pathway.
Addressing Cell-Type and Context-Specific Variability in UPR Activation
The Unfolded Protein Response (UPR) is a critical adaptive signaling network that maintains endoplasmic reticulum (ER) proteostasis under stress conditions. While the core UPR signaling pathways are well-defined, their activation dynamics and functional outcomes exhibit remarkable heterogeneity across different cell types and physiological contexts. This variability presents both challenges and opportunities for research and therapeutic development. The UPR comprises three major signaling branches—PERK, IRE1α, and ATF6—that collectively orchestrate a multifaceted response to ER stress [10] [118]. However, the induction magnitude and repertoire of UPR-regulated genes vary significantly by cell type, tissue, and the nature of the stressor [119]. This review examines the sources and implications of UPR heterogeneity while providing methodological frameworks for its systematic investigation in research and drug development.
Fundamental UPR Signaling Pathways
The UPR is initiated through three ER-transmembrane sensors that detect protein folding imbalances. Each branch activates distinct signaling cascades with both overlapping and unique functional outcomes.
The PERK-eIF2α-ATF4 Axis
PERK (PKR-like ER kinase) activation represents the most rapid response to ER stress. Upon dissociation from the chaperone BiP/GRP78, PERK dimerizes and autophosphorylates, then phosphorylates the eukaryotic initiation factor 2α (eIF2α) [10] [118]. This phosphorylation event globally attenuates protein translation to reduce the ER's protein-folding load while selectively promoting the translation of specific mRNAs, including that of the transcription factor ATF4 [10] [45]. ATF4 subsequently regulates genes involved in amino acid metabolism, antioxidant responses, and apoptosis. Under prolonged stress, ATF4 induces the expression of the pro-apoptotic factor CHOP (C/EBP homologous protein), shifting the UPR from adaptive to pro-apoptotic signaling [118] [45].
The IRE1α-XBP1 Pathway
IRE1α (inositol-requiring enzyme 1 alpha) possesses both kinase and endoribonuclease activities. Upon ER stress, IRE1α oligomerizes and autophosphorylates, activating its RNase domain to execute a two-pronged RNA processing response [10] [118]. Its primary function involves the unconventional splicing of X-box binding protein 1 (XBP1) mRNA, resulting in a frameshift that produces the stable and active transcription factor XBP1s [10]. XBP1s regulates genes expanding ER folding capacity, lipid biosynthesis, and ER-associated degradation (ERAD). IRE1α also cleaves specific mRNAs and pre-miRNAs through Regulated IRE1-Dependent Decay (RIDD), reducing the protein-folding load on the ER [10].
The ATF6 Proteolytic Cascade
ATF6 (activating transcription factor 6) is a type II ER transmembrane protein released from BiP during ER stress. It translocates to the Golgi apparatus, where it undergoes proteolytic cleavage by S1P and S2P proteases [118]. This cleavage releases its cytosolic domain (pATF6), which functions as a potent transcription factor that activates genes encoding ER chaperones, foldases, and components of the ERAD machinery [10] [118].
The following diagram illustrates the core signaling pathways and their crosstalk:
Cell-Type Specific Signaling
Different cell types exhibit distinct UPR activation patterns based on their specialized functions. For example, pancreatic β-cells, which have extensive ER to handle proinsulin secretion, demonstrate heightened sensitivity to ER stress, with chronic activation leading to apoptosis and the onset of diabetes [119] [118]. In contrast, cancer cells often exploit the UPR to survive hostile tumor microenvironments characterized by hypoxia, nutrient deprivation, and oxidative stress [17] [45]. The UPR's role in cancer is particularly complex, as it can both inhibit tumorigenesis initially and promote advanced disease through adaptation to stress [17].
Context-Dependent Modulation
The UPR does not function in isolation but exhibits extensive crosstalk with other stress response pathways. The PERK branch interfaces with the integrated stress response (ISR) through eIF2α phosphorylation, while IRE1α signaling intersects with JNK and TRAF2 pathways that influence inflammatory responses and apoptosis [119] [45]. Additionally, the mitochondrial UPR (mtUPR) communicates with ER stress pathways through mitochondria-associated membranes (MAMs), creating an interorganelle dialogue that further contributes to response heterogeneity [17].
Table 1: Factors Contributing to UPR Heterogeneity Across Biological Contexts
| Factor | Impact on UPR Signaling | Representative Example |
|---|---|---|
| Cell Type & Function | Determines basal UPR activity and threshold for activation | Pancreatic β-cells show heightened PERK sensitivity; plasma cells exhibit robust IRE1α-XBP1 signaling [119] [118] |
| Nature & Duration of Stress | Influences branch-specific activation dynamics and cell fate decisions | Transient stress promotes adaptive UPR; chronic stress triggers apoptotic signaling through CHOP [118] [45] |
| Tissue Microenvironment | Modulates UPR through nutrient availability, oxygen tension, and cellular interactions | Tumor microenvironment exploits UPR for survival; neuronal activity patterns regulate UPR in brain [17] [46] |
| Interpathway Crosstalk | Creates signaling networks that diversify UPR outcomes | PERK-NRF2 coordination in oxidative stress; IRE1α-TRAF2-JNK in inflammation [119] [45] |
| Developmental & Physiological Status | Alters UPR capacity and signaling preferences | Aging reduces UPR efficacy; circadian rhythms influence ER proteostasis [119] [46] |
Methodological Approaches for Resolving UPR Heterogeneity
Advanced Reporter Systems
Traditional UPR monitoring methods often fail to capture the complexity of branch-specific activation across heterogeneous cell populations. The recently developed sUPRa (sensor of UPR activity) reporter addresses this limitation by employing a dual-color system that captures global UPR activation while controlling for cell-to-cell variability in reporter copy number [46]. sUPRa utilizes a short region (bases -195 to -9) of the mouse BiP promoter—which contains response elements for all three UPR branches—to drive expression of the fast-maturing green fluorescent protein mNeonGreen (mNG). A second constitutively active promoter (EF1α) drives expression of mScarlet (mSc), providing an internal reference that enables ratiometric quantification of UPR induction independent of transfection efficiency or cell size [46].
Proteomic and Transcriptomic Profiling
Bulk analysis methods often mask cell-to-cell variability in UPR activation. Single-cell RNA sequencing (scRNA-seq) enables resolution of UPR heterogeneity at the transcriptional level across individual cells within a population. For proteomic assessment, Data-Independent Acquisition Mass Spectrometry (DIA-MS) has demonstrated superior performance in detecting low-abundance UPR-regulated proteins compared to traditional Data-Dependent Acquisition (DDA) methods [119]. This approach enabled the identification of branch-specific UPR proteomic targets that revealed differential regulation of the XBP1s branch in BRAF-mutant melanoma cells treated with BRAF inhibitors [119].
Table 2: Key Research Reagent Solutions for UPR Heterogeneity Studies
| Reagent/Tool | Function/Application | Key Features & Considerations |
|---|---|---|
| sUPRa Reporter [46] | Global UPR activity monitoring with cellular resolution | Dual-color (mNG/mSc) ratiometric quantification; superior sensitivity to physiological UPR; identifies heterogeneous activation |
| Branch-Selective Cell Lines [119] | Specific activation of individual UPR branches without global UPR induction | Enables deconvolution of branch-specific contributions; identifies specialized functions of PERK, IRE1α, and ATF6 |
| DIA Mass Spectrometry [119] | Comprehensive quantification of UPR-regulated proteins | Enhanced detection of low-abundance UPR effectors; automated SP3-based sample preparation; compatible with high-throughput screening |
| Pharmacological Activators/Inhibitors | Selective manipulation of UPR branches | Thapsigargin (ER Ca²⁺ depletion); Tunicamycin (N-glycosylation inhibition); specific PERK/IRE1α inhibitors (e.g., GSK2606414, 4μ8C) |
| Branch-Specific Proteomic Targets [119] | Validated protein markers for specific UPR pathway activation | Enables screening of UPR branch activity across cell types, treatments, and disease conditions; reveals context-dependent UPR regulation |
Experimental Framework for Cell-Type Specific UPR Analysis
Workflow for Heterogeneity Resolution
A systematic approach to addressing UPR variability involves sequential characterization from population-level assessment to single-cell resolution:
Initial Screening: Utilize branch-specific reporters or pharmacological modulators to identify overall UPR activation patterns in response to specific stimuli across different cell types.
Proteomic/Transcriptomic Profiling: Employ DIA-MS or scRNA-seq to quantify UPR branch activity and identify differentially regulated targets using validated branch-specific markers [119].
Functional Validation: Implement genetic (siRNA, CRISPR) or pharmacological approaches to selectively inhibit specific UPR branches and assess functional consequences on cellular outcomes.
Temporal Dynamics Assessment: Monitor UPR activation at multiple timepoints to distinguish adaptive versus terminal UPR signaling, as the same branch can promote survival initially but trigger apoptosis during prolonged activation [118] [45].
Data Interpretation Considerations
When analyzing cell-type and context-specific UPR responses, researchers should consider several key aspects: First, baseline ER capacity differs substantially across cell types, influencing the threshold for UPR activation. Second, non-canonical UPR functions continue to be discovered, including roles in lipid metabolism, inflammation, and differentiation [10] [17]. Third, the temporal dimension of UPR signaling critically determines functional outcomes, with rapid, transient activation typically promoting adaptation while sustained signaling often initiates apoptosis [118].
The cell-type and context-specific variability in UPR activation represents both a challenge for therapeutic targeting and an opportunity for precision medicine approaches. The methodologies and frameworks outlined herein provide a roadmap for resolving this complexity through integrated experimental strategies. As research continues to illuminate the diverse manifestations of ER stress across physiological and pathological contexts, embracing rather than ignoring this heterogeneity will be essential for developing effective UPR-targeted therapies for cancer, neurodegenerative diseases, metabolic disorders, and other conditions linked to proteostasis disruption.
The unfolded protein response (UPR) represents a critical adaptive signaling network activated by endoplasmic reticulum (ER) stress. While researchers often monitor individual UPR markers for convenience, this approach provides an incomplete and potentially misleading picture of ER homeostasis. This technical review examines why a multi-parameter analytical framework is essential for accurate UPR assessment. We synthesize evidence demonstrating that cell fate decisions under ER stress depend on the integrated signaling dynamics across all three UPR branches—IRE1, PERK, and ATF6—rather than the activation state of any single pathway. The article provides comprehensive methodologies for simultaneous UPR branch monitoring, detailed visualization of pathway interconnections, and standardized experimental approaches to overcome the limitations of single-marker analysis in ER stress research and drug development.
The endoplasmic reticulum serves as the primary intracellular organelle for protein synthesis, folding, and post-translational modifications. An imbalance between the protein-folding load and the ER's processing capacity leads to the accumulation of unfolded or misfolded proteins, a condition termed ER stress [2] [3]. To counteract this threat, cells activate the unfolded protein response (UPR), a sophisticated signaling network orchestrated by three ER-transmembrane sensors: inositol-requiring enzyme 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [2] [120] [3].
Under non-stress conditions, these sensors remain inactive through association with the chaperone protein BiP/GRP78 [3] [121]. The accumulation of unfolded proteins triggers BiP dissociation, initiating distinct signaling cascades from each sensor aimed at restoring proteostasis through multiple mechanisms, including translational attenuation, ER chaperone upregulation, and ER-associated degradation (ERAD) [120] [3]. However, under severe or prolonged stress, the UPR transitions from adaptive to pro-apoptotic signaling [120] [3].
The fundamental challenge in UPR monitoring stems from the network's dynamic, branch-specific signaling patterns and context-dependent outcomes. This technical guide establishes why measuring a single UPR marker provides insufficient evidence for conclusive ER stress assessment and outlines comprehensive methodological approaches for accurate pathway evaluation.
The Interconnected UPR Signaling Network
The Three Branches of the UPR
PERK-eIF2α-ATF4 Pathway Upon activation, PERK oligomerizes and autophosphorylates, then phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α) [120] [3]. This phosphorylation event globally attenuates protein translation to reduce the ER's protein-folding burden while simultaneously promoting the translation of select mRNAs, including that of activating transcription factor 4 (ATF4) [3]. ATF4 subsequently upregulates genes involved in amino acid metabolism, antioxidant response, and ER stress-induced apoptosis through the transcription factor CHOP (CCAAT/-enhancer-binding protein homologous protein) [3].
IRE1-XBP1 Pathway IRE1, the most conserved UPR sensor, possesses both kinase and endoribonuclease activities [3]. Upon ER stress, IRE1 oligomerizes and autophosphorylates, activating its RNase domain [120] [3]. Active IRE1 catalyzes the unconventional splicing of X-box binding protein 1 (XBP1) mRNA, removing a 26-nucleotide intron and producing a frameshift that generates the potent transcription factor spliced XBP1 (XBP1s) [122] [3]. XBP1s drives the expression of genes encoding ER chaperones, lipid biosynthesis enzymes, and ERAD components [120] [3].
ATF6 Pathway ATF6 is a type II transmembrane protein that translocates to the Golgi apparatus under ER stress conditions [120] [3]. There, it undergoes proteolytic cleavage by site-1 and site-2 proteases (S1P and S2P), releasing its cytosolic domain (ATF6f) [3]. This fragment functions as a transcription factor that enters the nucleus and enhances the expression of ER chaperones and XBP1 [3].
Integrated UPR Pathway Signaling
The following diagram illustrates the complex interactions and regulatory relationships between the three major UPR branches:
Figure 1: UPR Signaling Network Interconnections. This diagram illustrates the three major UPR branches and their complex interactions. The pathways initiate with BiP dissociation from sensors and culminate in either adaptive restoration of proteostasis or apoptosis induction. Note the cross-regulatory mechanisms (dashed lines) and feedback loops (dotted lines) that necessitate multi-parameter monitoring.
Key Limitations of Single-Marker Analysis
Temporal Dynamics and Signaling Heterogeneity
Single-timepoint measurement of any individual UPR marker fails to capture the critical temporal dynamics that determine cellular outcomes under ER stress conditions. Research using live-cell imaging of single cells expressing fluorescent UPR reporters has demonstrated that cell survival decisions depend on the relative timing and intensity of signaling through multiple UPR branches rather than the activation state of any single pathway [122].
Specifically, studies monitoring IRE1-mediated XBP1 splicing and PERK-mediated ATF4 translation in neural cells revealed that:
- An early onset and subsequent attenuation of XBP1 splicing correlates with cytoprotective outcomes [122]
- A slow rate of ATF4 translation combined with late re-initiation of general translation associates with cellular resistance to ER stress-induced apoptosis [122]
- Silencing IRE1 expression promotes early cell death, whereas PERK silencing primarily increases sensitivity to ER stressors at early timepoints without affecting overall cell death levels [122]
These findings indicate that the kinetic signature of UPR activation, rather than simple pathway engagement, determines cell fate. This temporal dimension is completely obscured when measuring a single marker at a single timepoint.
Branch-Specific Functional Outcomes
Each UPR branch regulates distinct aspects of the stress response with different functional consequences:
Table 1: Branch-Specific Functions of UPR Pathways
| UPR Branch | Primary Adaptive Functions | Pro-apoptotic Functions | Key Output Markers |
|---|---|---|---|
| PERK | Translational attenuation, antioxidant response, autophagy induction | CHOP-mediated apoptosis, GADD34-mediated dephosphorylation | p-eIF2α, ATF4, CHOP, GADD34 |
| IRE1 | ER chaperone production, ERAD component synthesis, lipid biosynthesis | RIDD-mediated decay of mRNAs, JNK activation via TRAF2 | XBP1 splicing, RIDD targets, JNK phosphorylation |
| ATF6 | ER chaperone transcription, XBP1 mRNA upregulation | Limited direct pro-apoptotic role | Cleaved ATF6, GRP78/BiP, HRD1 |
The table illustrates how each branch contributes both adaptive and pro-apoptotic signaling, creating a complex decision-making network where measuring only one marker provides insufficient information for predicting functional outcomes.
Cell-Type and Context-Dependent Signaling
UPR signaling exhibits remarkable cell-type specificity and contextual variation. For instance, in immune cells, the requirement for specific UPR branches varies considerably:
- B cells require IRE1-XBP1 signaling for differentiation into antibody-secreting plasma cells [121]
- T cells depend on PERK-eIF2α-ATF4 for Th2 differentiation and XBP1 for Th17 differentiation [121]
- Macrophages utilize IRE1α for NLRP3 inflammasome activation and inflammatory polarization [2]
- Dendritic cells employ different UPR branches for cytokine production depending on the pathogen-associated molecular pattern encountered [2]
This cell-type specificity means that a marker indicating adaptive signaling in one cellular context might signify pathological activation in another.
Feedback Regulation and Pathway Cross-Talk
The UPR contains multiple feedback mechanisms that create non-linear signaling dynamics:
- Negative feedback: GADD34, induced by CHOP, promotes eIF2α dephosphorylation to restore translation [3]
- Positive feedback: ATF6 enhances XBP1 transcription, creating amplification loops [3]
- Pathway integration: IRE1 signaling can degrade specific mRNAs through RIDD (regulated IRE1-dependent decay), including those encoding other UPR components [122]
These regulatory interactions mean that UPR branches do not function independently but form an integrated network with emergent properties that cannot be deduced from single-pathway measurements.
Comprehensive Experimental Approaches for UPR Assessment
Multi-Parameter Experimental Workflow
The following diagram outlines a standardized experimental workflow for comprehensive UPR assessment that addresses the limitations of single-marker approaches:
Figure 2: Comprehensive UPR Assessment Workflow. This experimental framework emphasizes simultaneous multi-branch monitoring, functional outcome correlation, and integrated data interpretation to overcome the limitations of single-marker analysis.
Standardized Methodologies for Multi-Branch UPR Monitoring
Simultaneous IRE1 and PERK Pathway Live-Cell Imaging
- Reporter Construction: Generate stable cell lines expressing:
- IRE1 reporter: Fluorescent protein-based XBP1 splicing reporter (e.g., XBP1-venus)
- PERK reporter: Fluorescent protein-based ATF4 translation reporter (e.g., ATF4-mCherry)
- Imaging Protocol: Perform time-lapse live-cell microscopy following ER stress induction
- Quantitative Analysis: Measure kinetics of reporter activation (time to onset, maximum intensity, duration)
- Data Interpretation: Correlate IRE1 and PERK signaling dynamics with cell survival/death outcomes [122]
Multi-Parameter Molecular Profiling
- Transcriptional Analysis: qPCR array measuring:
- IRE1 targets: EDEM1, ERdj4, HEDJ, p58IPK
- PERK targets: CHOP, GADD34, ERO1α
- ATF6 targets: GRP78/BiP, GRP94, HRD1, XBP1
- Protein Analysis: Western blotting for:
- Phosphorylated proteins: p-IRE1, p-PERK, p-eIF2α
- Transcription factors: ATF4, XBP1s, cleaved ATF6
- Effector proteins: CHOP, GRP78/BiP
- Functional Assays:
- ER folding capacity: Secreted protein analysis
- ERAD activity: Proteasome-sensitive protein degradation
- Apoptotic commitment: Caspase-3/7 activation, Annexin V staining
Research Reagent Solutions for Comprehensive UPR Analysis
Table 2: Essential Research Reagents for Multi-Parameter UPR Assessment
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin (N-glycosylation inhibitor), Thapsigargin (SERCA inhibitor), Brefeldin A (Golgi disruptor) | Controlled ER stress induction | Use multiple concentrations; validate time courses; include vehicle controls |
| Pathway Inhibitors | PERK: GSK2606414; IRE1: STF-083010, 4μ8c; ATF6: Ceapins | Pathway-specific functional validation | Assess specificity; potential off-target effects; use multiple inhibitors when possible |
| Reporter Systems | XBP1 splicing reporters (e.g., pXBP1-ADFP), ATF4 translation reporters, GRP78 promoter reporters | Live-cell imaging and kinetic analysis | Validate specificity; consider dual-reporter systems; account for reporter burden |
| Antibodies | p-eIF2α, total eIF2α, ATF4, CHOP, XBP1s, GRP78/BiP, cleaved ATF6 | Protein-level pathway assessment | Validate species reactivity; optimize phosphorylation conditions |
| qPCR Assays | XBP1 splicing assays, UPR target gene panels | Transcriptional output measurement | Design spanning exon-exon junctions; include reference genes |
Quantitative Framework for UPR Data Interpretation
Temporal Classification of UPR Signaling States
Based on single-cell imaging studies [122], UPR activation dynamics can be categorized into distinct temporal patterns that predict cellular outcomes:
Table 3: Temporal Patterns of UPR Activation and Cellular Outcomes
| UPR Signaling Pattern | IRE1-XBP1 Dynamics | PERK-ATF4 Dynamics | Cellular Outcome | Experimental Evidence |
|---|---|---|---|---|
| Adaptive Resolution | Early onset (<4h), rapid attenuation by 8h | Moderate onset, declining by 12-16h | Cell survival, restored proteostasis | [122] |
| Chronic Adaptation | Sustained intermediate activation | Low but persistent activation | Survival with reduced function, altered secretion | [122] [121] |
| Terminal Stress | Rapid strong activation followed by abrupt decline | Progressive increase without attenuation | Apoptosis initiation | [122] [3] |
| Failed Adaptation | Delayed or minimal activation | Sustained high-level activation | Apoptosis after prolonged stress | [122] |
Branch Activation Ratio Calculations
For quantitative UPR assessment, researchers can calculate branch activation ratios that provide more predictive value than individual marker levels:
IRE1:PERK Adaptive Ratio = (XBP1s target gene expression) / (CHOP expression)
Interpretation guidelines:
- Ratio > 2.0: Predominantly adaptive signaling
- Ratio 0.5-2.0: Mixed adaptive/apoptotic signaling
- Ratio < 0.5: Predominantly apoptotic signaling
Validation approach: Correlate ratio values with direct viability measures and functional ER capacity assays.
Comprehensive UPR assessment requires moving beyond single-marker analysis to embrace multi-parameter, time-resolved monitoring approaches. The experimental frameworks presented in this technical review provide standardized methodologies for capturing the dynamic interplay between UPR branches that ultimately determines cellular fate under ER stress conditions. By adopting these integrated assessment strategies, researchers can generate more meaningful data on UPR activation states, improve reproducibility across studies, and develop more accurate predictive models for therapeutic targeting of ER stress in disease contexts.
The complexity of UPR signaling necessitates this comprehensive approach, as single markers cannot capture the network properties, temporal dynamics, or cell-type specificity that are fundamental to understanding ER stress pathophysiology and developing effective interventions.
UPR in Pathophysiology and Therapy: Validating Its Role Across Disease Landscapes
The Unfolded Protein Response (UPR) is an evolutionarily conserved signaling network that maintains proteostasis within the endoplasmic reticulum (ER). In oncogenesis, this pathway assumes a complex dual role: it initially functions as an adaptive mechanism promoting tumor cell survival under stress but can ultimately trigger apoptosis when stress becomes severe or prolonged [123] [10]. Cancer cells, existing in microenvironments characterized by hypoxia, nutrient deprivation, and metabolic stress, experience persistent Endoplasmic Reticulum (ER) stress [17]. Consequently, they co-opt the UPR not merely as a stress-response pathway but as a critical facilitator of malignant progression, therapeutic resistance, and immune evasion [123] [17]. Understanding this dichotomous nature—where the UPR serves as both a guardian of cell integrity and an agent of cell death—is paramount for developing novel cancer therapeutics that exploit this vulnerability. This review synthesizes current knowledge on the UPR's multifaceted functions in cancer, detailing its mechanisms, context-dependent outcomes, and emerging strategies for its therapeutic targeting.
Molecular Mechanisms of the UPR
The UPR is coordinated by three ER-transmembrane sensors: IRE1α, PERK, and ATF6. Under non-stress conditions, these sensors are largely inactive, bound to the chaperone BiP (GRP78). The accumulation of unfolded proteins leads to BiP dissociation, triggering the activation of each arm [123] [3].
Diagram 1: The Tripartite UPR Signaling Network. The three ER stress sensors (IRE1α, PERK, ATF6) initiate adaptive signaling to restore proteostasis. Under sustained stress, pro-apoptotic pathways, primarily via CHOP and JNK, are activated, leading to cell death.
The IRE1α-XBP1 Axis
IRE1α, the most evolutionarily conserved branch, possesses both kinase and endoribonuclease activities. Upon activation, it catalyzes the unconventional splicing of XBP1 mRNA, producing a potent transcription factor (XBP1s) that drives the expression of genes involved in ER-associated degradation (ERAD), lipid synthesis, and chaperone production [123] [10]. Under prolonged ER stress, IRE1α can also initiate apoptosis through its interaction with TRAF2, activating the ASK1/JNK pathway [123]. Additionally, IRE1α's RNase activity can degrade a subset of mRNAs and miRNAs via Regulated IRE1-Dependent Decay (RIDD), further reducing the protein-folding load on the ER [10].
The PERK-eIF2α-ATF4/CHOP Axis
PERK activation leads to the immediate phosphorylation of eukaryotic Initiation Factor 2α (eIF2α). This phosphorylation globally attenuates cap-dependent protein translation, reducing the influx of new proteins into the stressed ER. However, it simultaneously promotes the translation of select mRNAs, including that of Activating Transcription Factor 4 (ATF4) [123] [3]. ATF4 upregulates genes involved in amino acid metabolism, antioxidant responses, and autophagy. Critically, persistent PERK signaling and ATF4 activation induce the expression of the C/EBP Homologous Protein (CHOP), a key mediator of ER stress-induced apoptosis. CHOP suppresses anti-apoptotic BCL-2 while inducing pro-apoptotic proteins like BIM and PUMA [123] [17].
The ATF6 Signaling Cascade
ATF6 is a type II transmembrane protein that, upon ER stress, translocates to the Golgi apparatus. There, it is cleaved by Site-1 Protease (S1P) and Site-2 Protease (S2P), releasing its cytosolic N-terminal fragment (ATF6f) [123]. This fragment acts as a transcription factor, migrating to the nucleus to enhance the expression of ER chaperones (like BiP) and XBP1, thereby collaborating with the IRE1α arm to expand the ER's folding capacity [123] [3].
The UPR as a Driver of Tumorigenesis and Therapy Resistance
Cancer cells exploit the adaptive UPR to thrive in hostile microenvironments and resist therapeutic insults. The following table summarizes key pro-tumorigenic processes supported by UPR signaling.
Table 1: Oncogenic Processes Facilitated by UPR Signaling
| UPR Arm | Pro-Tumorigenic Function | Mechanism of Action | Associated Cancers |
|---|---|---|---|
| IRE1α-XBP1 | Metabolic Reprogramming, Angiogenesis, Metastasis | XBP1s induces lipid synthesis, VEGF; RIDD degrades metastasis suppressors [17]. | Breast, Triple-Negative Breast Cancer [17] |
| PERK-ATF4 | Angiogenesis, Immune Evasion, Oxidative Stress Management | ATF4 induces VEGF; Promotes PD-L1 expression; Activates amino acid transport & antioxidant synthesis [123] [17]. | Sarcomas, Glioblastoma [123] |
| ATF6 | Apoptotic Resistance, Metastasis | Enhances chaperone expression & promotes pro-survival signaling; Calreticulin expression linked to invasion [17]. | Various solid tumors [17] |
| UPR General (e.g., BiP/GRP78) | Therapy Resistance, Immune Modulation | Upregulated BiP inhibits apoptosis; UPR creates immunosuppressive TME by modulating macrophages and T-cells [17] [3]. | Ovarian Cancer, Glioma, Prostate Cancer [17] [124] |
Metabolic Adaptation and Angiogenesis
The UPR enables tumors to overcome nutrient and oxygen deprivation. The PERK-ATF4 axis is a crucial regulator of redox homeostasis and amino acid metabolism [123]. Furthermore, both PERK and IRE1α contribute to angiogenesis by inducing Hypoxia-Inducible Factors (HIFs) and VEGF, ensuring an adequate blood supply [17].
Immune Evasion
UPR activation fosters an immunosuppressive Tumor Microenvironment (TME). For instance, PERK signaling can upregulate the expression of PD-L1 on tumor cells, facilitating T-cell exhaustion [17]. UPR activation in tumor-associated macrophages (TAMs) can polarize them toward a pro-tumorigenic phenotype, further suppressing anti-tumor immunity [17].
Therapy Resistance
The UPR is a cornerstone of intrinsic and acquired resistance to chemotherapy, targeted therapy, and radiotherapy. By enhancing protein-folding capacity, promoting autophagy, upregulating anti-apoptotic proteins, and managing oxidative stress, the UPR allows cancer cells to withstand therapeutic insults [17] [124]. For example, in ovarian cancer, basal UPR upregulation is associated with platinum resistance, and its further induction can push cells toward apoptosis, revealing a therapeutic vulnerability [124].
Experimental Validation: Methodologies and Reagents
Validating the UPR's role requires a multi-faceted approach, from in vitro models to clinical correlative studies.
In Vitro Functional Assays
Cell Viability and Death under ER Stress:
- Protocol: Seed cancer cells in 96-well plates. Treat with ER stress inducers (e.g., Tunicamycin, Thapsigargin) or UPR-targeting compounds (e.g., ONC201) across a dose range. After 48-72 hours, assess viability using MTT or MTS assays, which measure metabolic activity [124]. Confirm cytotoxic effects via Trypan Blue Exclusion for direct cell counting [124].
- Apoptosis Detection: Use flow cytometry with Annexin V/Propidium Iodide (PI) staining to distinguish early apoptotic (Annexin V+/PI-) and late apoptotic/necrotic (Annexin V+/PI+) populations after drug treatment [124].
- Migration and Invasion: Perform Wound Healing (Scratch) Assays. Create a scratch in a confluent cell monolayer, treat with the compound of interest, and monitor wound closure over 24-48 hours using time-lapse microscopy to evaluate anti-migratory effects [124].
Gene Expression Analysis:
- qRT-PCR for UPR Target Genes: Extract total RNA and synthesize cDNA. Use primers specific for UPR markers (e.g., XBP1s, ATF4, CHOP, BiP) for quantitative analysis. The splicing of XBP1 can be detected using specific primers or by resolving PCR products on agarose gels [125] [124].
- Western Blotting for Protein-Level Validation: Lyse cells in RIPA buffer. Separate proteins by SDS-PAGE, transfer to a membrane, and probe with antibodies against key UPR proteins (e.g., p-eIF2α, ATF4, CHOP, XBP1s, ATF6f) and apoptosis markers (e.g., cleaved PARP, BIM) [124]. β-actin serves as a loading control.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for UPR and Cancer Research
| Reagent / Tool | Function / Target | Key Application in UPR Research |
|---|---|---|
| Tunicamycin | N-linked Glycosylation Inhibitor | Classic ER stress inducer; used to activate all three UPR arms and study UPR kinetics [125]. |
| Thapsigargin | Sarco/Endoplasmic Reticulum Ca²⁺ ATPase (SERCA) Inhibitor | Induces ER stress by disrupting Ca²⁺ homeostasis; used to study UPR and calcium-mediated apoptosis. |
| ONC201 (Dordaviprone) | DRD2 Antagonist / ClpP Activator | Investigational compound that induces the Integrated Stress Response (ISR) and UPR; used to study ER stress-mediated apoptosis in cancer [126] [124]. |
| GSK2606414 | PERK Inhibitor | Potent and selective PERK kinase inhibitor; used to delineate the specific role of the PERK arm in tumor survival and therapy resistance [17]. |
| 4μ8C | IRE1α RNase Inhibitor | Selective inhibitor of IRE1α's RNase domain; used to block XBP1 splicing and RIDD, probing IRE1α's role in tumorigenesis [17]. |
| Anti-CHOP Antibody | Transcription Factor CHOP | Detects CHOP protein expression via Western Blot or IHC; a key marker for the terminal, pro-apoptotic UPR [124]. |
| Anti-XBP1s Antibody | Spliced XBP1 (Transcription Factor) | Specifically detects the active, spliced form of XBP1; crucial for monitoring IRE1α arm activation [17]. |
Therapeutic Targeting of the UPR in Oncology
The dependency of many tumors on UPR signaling creates a therapeutic window. Strategies include inhibiting pro-survival UPR arms or hyperactivating the UPR to lethal levels.
Diagram 2: Strategic Approaches for Targeting the UPR in Cancer. Therapeutic interventions either inhibit pro-survival UPR signaling to dismantle tumor adaptation or hyperactivate the UPR to push cells beyond their proteostatic capacity and into apoptosis.
UPR Inhibitors
- PERK Inhibitors: Compounds like GSK2606414 block PERK kinase activity, disrupting adaptive translation repression and ATF4-mediated survival signaling, thereby sensitizing tumors to ER stress [17].
- IRE1α Modulators: The RNase inhibitor 4μ8C blocks XBP1 splicing and RIDD. This can inhibit the production of pro-tumorigenic XBP1s and potentially lead to the accumulation of misfolded proteins [17].
- GRP78/BiP Targeting: The chaperone BiP is frequently overexpressed in tumors. Small molecules like HA15 or the monoclonal antibody PAT-SM6 bind to BiP, inhibiting its pro-survival function and inducing ER stress-mediated apoptosis [17].
UPR Inducers and Combination Therapy
An alternative strategy is to induce intolerable levels of ER stress. The imipridone ONC201, an FDA-approved agent for H3K27M-mutant glioma, activates the integrated stress response (ISR) and UPR, leading to ATF4/CHOP upregulation and apoptosis in various cancers, including ovarian and sarcoma [126] [124]. Its efficacy is often enhanced in combination with standard chemotherapy (e.g., paclitaxel) or targeted agents, as shown in preclinical models of platinum-resistant ovarian cancer [124]. Combining UPR modulators with immunotherapy is also a promising frontier, given the UPR's role in regulating the immune TME [17].
The UPR represents a paradigm of cellular stress adaptation that is fundamentally co-opted in oncogenesis. Its dual role—facilitating survival under transient stress yet committing the cell to death upon failure to restore proteostasis—presents both a challenge and a unique therapeutic opportunity. Future research must focus on dissecting the context-dependent functions of individual UPR arms across different cancer types and genetic backgrounds [17]. A critical challenge is the potential on-target toxicity of UPR inhibition in normal secretory tissues. The development of predictive biomarkers—such as high XBP1 splicing or CHOP expression—will be essential for selecting patients most likely to benefit from UPR-directed therapies [123] [17]. As novel UPR modulators enter clinical trials, often in rational combinations, the goal remains to strategically manipulate this intricate network, pushing malignant cells across the fragile threshold from survival to self-destruction.
The endoplasmic reticulum (ER) unfolded protein response (UPR) has long been a cornerstone of cellular stress research, representing a critical mechanism for maintaining proteostasis. However, an equally vital but less explored pathway operates within the mitochondria—the mitochondrial unfolded protein response (mtUPR). This mitochondrial stress response represents an evolutionarily conserved adaptive mechanism that safeguards mitochondrial proteostasis and function under diverse stress conditions. While the ER UPR addresses proteostatic imbalances within the endoplasmic reticulum, the mtUPR specifically monitors and responds to perturbations in the mitochondrial protein-folding environment [127]. The significance of mtUPR extends across numerous pathological contexts, particularly in neurological disorders and cancer, where its targeted regulation shows promise for therapeutic intervention [127] [128].
Beyond its organelle-specific functions, the mtUPR does not operate in isolation but participates in extensive interorganellar communication, particularly with the ER. This crosstalk creates an integrated cellular stress response network that coordinates metabolic adaptation, redox homeostasis, and cell fate decisions under challenging conditions [129] [130]. The physical and functional coupling between mitochondria and ER occurs through specialized structures known as mitochondria-associated membranes (MAMs), which serve as platforms for lipid transfer, calcium signaling, and molecular communication [131]. Understanding this sophisticated dialogue between organelles provides novel insights into cellular homeostasis and reveals potential therapeutic targets for complex diseases characterized by proteostatic dysfunction.
Molecular Mechanisms of mtUPR Signaling
Core Components and Activation Triggers
The mitochondrial unfolded protein response initiates when the protein-folding capacity within mitochondria becomes overwhelmed by damaged or misfolded proteins. This overload typically results from various stressors, including mitochondrial reactive oxygen species (mtROS) that damage mitochondrial proteins, mtDNA mutations that disrupt electron transport chain complexes, and metabolic perturbations that challenge energy homeostasis [128]. Under physiological conditions, mitochondrial chaperones and proteases maintain proteostasis by facilitating proper protein folding and degrading damaged proteins. However, when misfolded proteins accumulate beyond handling capacity, they trigger the activation of the mtUPR signaling cascade [127].
Central to this response are several key molecular players. Mitochondrial heat shock protein 70 (mtHsp70) and other chaperones facilitate the import and refolding of proteins within the mitochondrial matrix [127]. Proteases such as caseinolytic protease P (CLPP) function to degrade irreparably damaged proteins, preventing toxic aggregation [127] [128]. The activation mechanism involves the transcription factor activating transcription factor associated with stress 1 (ATFS-1) in C. elegans, while mammalian systems employ a more complex regulatory network involving ATF5, ATF4, and CHOP [127]. When mitochondrial protein import is compromised due to folding stress, ATFS-1 (or its mammalian counterparts) escapes mitochondrial degradation and translocates to the nucleus, where it activates genes encoding mitochondrial chaperones, proteases, and antioxidant defenses [127] [128].
Comparative Analysis: mtUPR versus ER UPR
While both unfolded protein responses share the fundamental purpose of maintaining proteostasis, their signaling mechanisms and functional outcomes exhibit significant differences, as outlined in the table below.
Table 1: Comparative analysis of mtUPR versus ER UPR
| Feature | mtUPR | ER UPR |
|---|---|---|
| Primary Inducers | mtROS, mtDNA mutations, protein import defects | Glucose deprivation, calcium flux, redox imbalance, glycosylation defects |
| Key Sensors | ATFS-1 (C. elegans), ATF5/ATF4/CHOP (mammals) | PERK, IRE1α, ATF6 |
| Major Effectors | Mitochondrial chaperones (mtHsp70), proteases (CLPP) | ER chaperones (BiP/GRP78), ERAD components |
| Transcriptional Regulators | ATFS-1, ATF5, CHOP | XBP1s, ATF4, ATF6f, CHOP |
| Primary Functions | Restore mitochondrial proteostasis, enhance ROS detoxification | Increase ER folding capacity, reduce protein load, enhance ERAD |
| Crosstalk Mechanisms | Integrated stress response (ISR), calcium signaling, redox signaling | PERK-eIF2α signaling, calcium release, redox signaling |
The UPR branches exhibit distinct yet complementary functions. The ER UPR employs three transmembrane sensors—PERK, IRE1α, and ATF6—that detect misfolded proteins in the ER lumen and initiate signaling cascades to restore folding capacity [2] [3]. IRE1α splices XBP1 mRNA to generate the active transcription factor XBP1s, which upregulates ER chaperones and components of ER-associated degradation (ERAD) [3]. PERK phosphorylates eIF2α to attenuate global protein synthesis while selectively promoting ATF4 translation, which activates genes involved in amino acid metabolism, antioxidant response, and apoptosis [3]. ATF6 traffics to the Golgi upon ER stress, where it is cleaved to release its cytosolic domain (ATF6f), which migrates to the nucleus to enhance expression of chaperones and ERAD components [3].
Despite their distinct activation mechanisms and organelle specificity, the mtUPR and ER UPR converge through shared transcription factors and signaling nodes. CHOP, for instance, is activated by both mitochondrial and ER stress and can promote apoptosis under severe or prolonged stress conditions [127] [3]. The phosphorylation of eIF2α represents another critical integration point, as it occurs in response to diverse stresses and regulates translation in both compartments [127]. This sophisticated network enables cells to mount a coordinated response to proteostatic challenges that often simultaneously affect multiple organelles.
Experimental Methodologies for mtUPR Research
Established Protocols for mtUPR Induction and Detection
Investigating mtUPR activation requires carefully controlled experimental conditions and specific methodological approaches. Researchers commonly employ both pharmacological and genetic tools to induce mitochondrial stress and subsequently measure specific readouts of pathway activation.
Pharmacological Induction Protocols:
- Complex I Inhibitors (Rotenone): Treat cells with 100-500 nM rotenone for 6-24 hours to induce mitochondrial electron transport chain dysfunction and increase mtROS production [128].
- Complex III Inhibitors (Antimycin A): Use 1-10 µM antimycin A for 6-12 hours to disrupt electron flow, promoting superoxide generation from Complex III [128].
- xeno-nucleic acids (XNAs): Apply 1-5 µM of specific XNAs for 12-24 hours to inhibit mitochondrial translation and induce proteostatic stress [127].
Key Detection Methodologies:
- Quantitative PCR for mtUPR Marker Genes: Measure transcript levels of canonical mtUPR markers including HSP60, HSP10, mtDAMP, and CLPP using SYBR Green or TaqMan protocols with normalization to housekeeping genes [127]. Primers should be designed to span intron-exon boundaries where applicable.
- Western Blot Analysis of UPR Components: Resolve 20-50 µg of mitochondrial or whole-cell extracts on 4-12% Bis-Tris gels, transfer to PVDF membranes, and probe with antibodies against ATF5, HSP60, CHOP, or phosphorylated eIF2α [127] [3]. Proper mitochondrial fractionation is critical for assessing compartment-specific responses.
- Immunofluorescence and Microscopy: Fix cells with 4% paraformaldehyde, permeabilize with 0.1% Triton X-100, and incubate with primary antibodies against mtUPR components followed by fluorophore-conjugated secondary antibodies. Confocal microscopy enables subcellular localization assessment [127].
Advanced Technical Approaches
Recent technological advances have significantly expanded our ability to investigate mtUPR dynamics and interorganellar communication with unprecedented resolution.
High-Throughput Omics Technologies:
- Single-Cell RNA Sequencing (scRNA-seq): This approach reveals cell-type-specific mtUPR activation patterns and heterogeneous stress responses within complex tissues [127]. The standard protocol involves single-cell suspension preparation, droplet-based partitioning, barcoded cDNA library preparation, and sequencing followed by bioinformatic analysis of mtUPR-related gene signatures.
- Spatial Transcriptomics: This method maps regions of high mtUPR activity within tissue architecture, preserving spatial context that is lost in conventional sequencing approaches [127]. Fresh-frozen tissue sections are mounted on patterned capture slides containing spatial barcodes, followed by sequencing and reconstruction of mtUPR gene expression patterns within histological contexts.
- Proteomic Analysis of MAM Fractions: Isolate mitochondria-associated membranes (MAMs) through differential centrifugation and Percoll gradient separation, then analyze protein composition using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify novel regulators of ER-mitochondria crosstalk [131].
Live-Cell Imaging and Dynamics:
- Super-Resolution Microscopy: Techniques such as STORM or STED microscopy visualize the dynamic interactions between ER and mitochondria at MAMs with nanometer-scale resolution, using specific markers for each organelle [127].
- FRET-Based Calcium Sensors: Genetically encoded reporters such as Cameleons enable real-time monitoring of calcium flux between ER and mitochondria, a key signaling modality in interorganellar communication [131] [130].
- Mitochondrial-Targeted ROS Sensors: Tools like MitoSOX Red or genetically encoded roGFP targeted to mitochondria allow quantitative assessment of mtROS production under stress conditions that trigger mtUPR [128].
Table 2: Essential research reagents for mtUPR and organelle crosstalk studies
| Reagent Category | Specific Examples | Primary Research Application |
|---|---|---|
| Chemical Inducers | Tunicamycin, Thapsigargin | Induce ER stress to study UPRER-mtUPR crosstalk [2] |
| mtUPR Activators | Rotenone, Antimycin A | Generate mtROS and trigger mtUPR [128] |
| Signal Inhibitors | STF-083010 (IRE1α inhibitor), GSK2606414 (PERK inhibitor) | Dissect specific UPR branch contributions [2] |
| Antibodies | Anti-HSP60, Anti-ATF5, Anti-CHOP, Anti-XBP1s | Detect mtUPR and UPRER components via WB/IF [127] [3] |
| Fluorescent Reporters | MitoTimer, MitoSOX, ER-GFP, Mito-DsRed | Visualize organelle architecture, dynamics, and stress [127] |
| Genetic Tools | ATFS-1-GFP fusion constructs, CLPP knockout cells | Monitor mtUPR activation and assess protease functions [127] [128] |
Interorganellar Crosstalk: ER-Mitochondria Communication
Structural and Functional Basis of Organelle Communication
The communication between endoplasmic reticulum and mitochondria occurs through specialized physical interfaces known as mitochondria-associated membranes (MAMs) or ER-mitochondria encounter structures (ERMES) [131] [130]. These dynamic membrane contact sites enable the direct exchange of lipids, calcium ions, and metabolic signals without resorting to vesicular transport through the cytosol. The distance between ER and mitochondrial membranes at these contact sites typically ranges from 10 to 80 nanometers, maintained by specific protein tethers that bridge the organelles [131].
Key molecular components facilitate this physical and functional coupling. Mitofusin 2 (Mfn2) localizes to both ER and mitochondrial membranes, serving as a critical tethering protein that regulates contact site formation and function [131]. The IP3R-GRP75-VDAC complex forms a molecular bridge that enables efficient calcium transfer from ER stores to the mitochondrial matrix, regulating energy production and cell death pathways [131]. FUN14 domain-containing protein 1 (FUNDC1), typically a mitophagy receptor, also participates in MAM regulation under stress conditions [131]. These specialized contact sites facilitate crucial cellular processes including lipid synthesis and transfer, calcium-mediated metabolic regulation, and coordinated stress signaling between the two organelles [131] [130].
The structural integrity of MAMs is essential for maintaining cellular homeostasis. Disruption of these contact sites, as observed in various disease states, impairs calcium transfer, lipid metabolism, and integrated stress responses. For instance, mutated REEP1, associated with hereditary spastic paraplegia, loses its ability to promote ER-mitochondria interactions, leading to defective contact sites and neuronal degeneration [130]. Similarly, caveolin-1 (CAV1) scaffolds regulate contact site abundance through the PKA-Drp1 signaling axis, influencing mitochondrial dynamics and function [130].
Integrated Stress Signaling Between Organelles
The communication between ER and mitochondria extends beyond structural coupling to encompass sophisticated signaling integration. During cellular stress, both organelles activate response pathways that exhibit significant crosstalk and coordination. The PERK branch of the UPRER serves as a novel molecular mediator of ER-mitochondria contact sites, regulating interorganellar crosstalk particularly during ROS-induced cell death [130]. Under glucose limitation, PERK activation remodels electron transport chain complexes without completely restructuring the mitochondrial proteome, demonstrating metabolic coordination between the compartments [130].
Calcium signaling represents a particularly crucial aspect of ER-mitochondria communication. Calcium release from ER stores through IP3 receptors is efficiently transferred to mitochondria through MAMs, where it regulates dehydrogenase activity and ATP production [131] [130]. However, under pathological conditions, excessive calcium transfer can trigger mitochondrial permeability transition pore opening and apoptosis. This calcium-mediated apoptosis is regulated by Bcl-2 family proteins that localize to both ER and mitochondria, demonstrating how cell fate decisions integrate signals from both organelles [131].
Redox signaling provides another key communication channel. Mitochondrial ROS production influences ER redox conditions, affecting disulfide bond formation and protein folding in the ER lumen. Conversely, ER stress can increase mitochondrial ROS production through multiple mechanisms, including calcium-mediated stimulation of mitochondrial metabolism and transcriptional upregulation of ERO-1α by CHOP, which promotes oxidative protein folding in the ER while generating hydrogen peroxide as a byproduct [3]. This bidirectional redox communication creates feedback loops that can either enhance cellular adaptation or promote apoptotic signaling depending on stress intensity and duration.
Diagram 1: ER-mitochondria crosstalk via MAMs. This diagram illustrates the bidirectional communication between endoplasmic reticulum (ER) and mitochondria occurring at mitochondria-associated membranes (MAMs), including calcium flux, lipid transfer, and redox signaling, as well as the crosstalk between their respective unfolded protein responses.
Pathophysiological Implications and Therapeutic Targeting
mtUPR in Disease Contexts
Dysregulation of mtUPR and interorganellar communication contributes significantly to various human diseases, particularly in tissues with high metabolic demands such as neurons and cancer cells. In neurological disorders including Alzheimer's disease, Parkinson's disease, and ischemic/reperfusion injury, mitochondrial dysfunction represents a critical pathological feature [127]. Neurons, with their exceptional metabolic requirements, particularly depend on mitochondrial ATP production via oxidative phosphorylation [127]. In these contexts, UPRmt activation serves as a compensatory mechanism that maintains mitochondrial proteostasis and energetic homeostasis under stress conditions [127]. Preclinical studies demonstrate that targeted regulation of UPRmt can effectively delay disease progression and improve functional outcomes, highlighting its therapeutic potential [127].
In cancer biology, UPRmt functions as a form of non-oncogene addiction that supports tumor growth and survival under metabolic stress [128]. Cancer cells with elevated mtROS production due to mitochondrial dysfunction rely on UPRmt activation to mitigate proteotoxic stress and maintain mitochondrial integrity [128]. Specific UPRmt components show altered expression in various cancers, with chaperones like HSP60 and proteases such as CLPP promoting tumor progression by alleviating mitochondrial proteotoxicity [128]. This dependency creates a therapeutic vulnerability, as inhibition of UPRmt components can selectively target cancer cells while sparing normal tissues with less mitochondrial stress.
The integrated stress response also plays crucial roles in metabolic diseases such as obesity and diabetes. In macrophages within adipose tissue, saturated fatty acids engage the IRE1α pathway to promote NLRP3 inflammasome activation and IL-1β secretion, contributing to insulin resistance [2]. Myeloid-specific deletion of IRE1α protects mice from diet-induced obesity and metabolic dysfunction by shifting macrophage polarization from pro-inflammatory M1 to anti-inflammatory M2 phenotypes [2]. Similarly, CHOP expression in adipocytes of obese mice drives local tissue inflammation by promoting M1 macrophage polarization, establishing a link between ER stress in one cell type and inflammatory responses in another [2].
Emerging Therapeutic Strategies
Targeting mtUPR and organelle crosstalk represents a promising frontier for therapeutic development across multiple disease contexts. Several strategic approaches are currently under investigation:
Small Molecule Modulators:
- IRE1α RNase Inhibitors: Compounds such as STF-083010 have shown efficacy in obese mouse models, ameliorating insulin resistance and reducing pro-inflammatory macrophage accumulation in adipose tissue [2].
- PERK Inhibitors: GSK2606414 and similar compounds block PERK signaling, reducing ER stress-mediated apoptosis in neurodegenerative models while potentially affecting mitochondrial function through altered MAM dynamics [3] [130].
- Antioxidant Approaches: Mitochondria-targeted antioxidants like MitoQ specifically reduce mtROS, potentially decreasing UPRmt activation by lowering the burden of oxidatively damaged proteins [128].
Gene Therapy and Precision Medicine:
- ATF5 Manipulation: As a central transcription factor in mammalian UPRmt, modulating ATF5 activity or expression represents a potential strategy for neurological disorders where mitochondrial proteostasis is compromised [127].
- MAM Stabilizers: Compounds that normalize ER-mitochondria contacts without causing pathological tethering could restore calcium signaling and lipid metabolism in diseases characterized by disrupted organelle communication [131] [130].
The development of these therapeutic approaches requires careful consideration of tissue-specific expression patterns, feedback regulation within stress response pathways, and the dual roles of many UPR components in both survival and apoptosis signaling. Future efforts will likely focus on achieving temporal and spatial precision in targeting these pathways to maximize therapeutic benefits while minimizing adverse effects.
Diagram 2: Therapeutic targeting strategies. This diagram outlines the relationship between specific disease contexts, their relevant molecular targets in UPR pathways, and the resulting therapeutic approaches under development.
The exploration beyond the ER to encompass mtUPR and interorganellar crosstalk represents a paradigm shift in our understanding of cellular proteostasis. Rather than existing as isolated compartments, organelles form dynamic communication networks that integrate stress signals and coordinate adaptive responses. The mtUPR has emerged as a crucial component of this network, maintaining mitochondrial function under diverse challenges while engaging in bidirectional communication with the ER UPR and other cellular stress response pathways.
Future research directions will likely focus on several key areas. First, understanding the spatiotemporal dynamics of organelle communication will require advanced imaging technologies and computational modeling to capture the rapid and localized nature of these interactions. Second, elucidating cell-type-specific variations in UPRmt signaling will be essential for developing targeted therapies that account for tissue-specific vulnerabilities. Third, exploring the metabolic integration between organelles during stress responses will provide insights into how bioenergetic demands shape proteostatic adaptations. Finally, translating basic discoveries about UPRmt and organelle crosstalk into clinical applications represents the ultimate frontier, with promising approaches already emerging for neurological disorders, cancer, and metabolic diseases.
As these research avenues expand, they will undoubtedly reveal new aspects of the sophisticated dialogue between organelles, further enriching our understanding of cellular homeostasis and providing novel therapeutic opportunities for complex diseases characterized by proteostatic dysfunction.
UPR as a Diagnostic and Prognostic Biomarker in Acute Leukemias and Solid Tumors
The unfolded protein response (UPR) represents an evolutionarily conserved cytoprotective signaling cascade essential for maintaining endoplasmic reticulum (ER) homeostasis under stress conditions [123] [4]. As a dynamic pathway that balances cell survival and apoptosis, the UPR has emerged as a critical modulator of cancer cell fate, with demonstrated value as diagnostic and prognostic biomarkers across diverse malignancies [132] [17]. Cancer cells experience significant endoplasmic reticulum stress due to the hypoxic, nutrient-deprived conditions of the tumor microenvironment coupled with their accelerated protein synthesis demands [123] [17]. This stress creates selective pressure for malignant cells to exploit the adaptive UPR to promote survival, growth, and therapeutic resistance [132] [4]. The three principal UPR sensors—inositol-requiring enzyme 1 (IRE1α), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor-6 (ATF6)—orchestrate a complex transcriptional program that enables tumor cells to withstand microenvironmental challenges [123] [2]. Recent research has revealed that UPR activation correlates strongly with aggressive phenotypes and poorer outcomes in multiple cancer types, positioning UPR-related genes (URGs) and signaling pathways as promising biomarkers for prognosis prediction and treatment selection [133] [132] [89]. This technical review comprehensively examines the application of UPR signaling components as diagnostic and prognostic biomarkers in acute leukemias and solid tumors, with particular emphasis on validated molecular signatures, experimental methodologies, and clinical applications.
Molecular Mechanisms of UPR Signaling
The mammalian UPR comprises three distinct signaling cascades mediated by ER transmembrane proteins: PERK (EIF2AK3), ATF6, and IRE1 (ERN1) [123] [4]. Under basal conditions, the ER chaperone HSPA5 (GRP78/BiP) binds these sensors' luminal domains, maintaining them in an inactive state. During ER stress, HSPA5 dissociates to bind misfolded proteins, initiating sensor activation through homodimerization and autophosphorylation (PERK, IRE1) or Golgi translocation (ATF6) [4].
The Three UPR Sensor Pathways
PERK-EIF2α-ATF4 Pathway: PERK activation initiates phosphorylation of eukaryotic translation initiation factor 2α (EIF2α), globally attenuating protein translation while selectively promoting translation of activating transcription factor 4 (ATF4) [123] [4]. ATF4 then activates processes including the antioxidant response, amino acid biosynthesis, and autophagy. Under prolonged stress, ATF4 induces the pro-apoptotic transcription factor DDIT3 (CHOP) [123].
IRE1-XBP1 Pathway: IRE1α, the most evolutionarily conserved UPR branch, possesses kinase and endoribonuclease activity. Activated IRE1 catalyzes unconventional splicing of XBP1 mRNA, generating a potent transcription factor (XBP1s) that drives expression of genes involved in ER biogenesis, protein folding, and degradation [123] [89]. Sustained IRE1 activation also triggers regulated IRE1-dependent decay (RIDD) of mRNAs and promotes apoptosis through TRAF2-ASK1-JNK signaling [123].
ATF6 Pathway: HSPA5 dissociation enables ATF6 translocation to the Golgi, where proteolytic cleavage by MBTPS1 and MBTPS2 liberates its cytosolic domain (ATF6f). This fragment functions as a potent transcription factor that upregulates ER chaperones, XBP1, and components of ER-associated degradation (ERAD) [123] [4].
Diagram Title: UPR Signaling Pathways in Cell Fate Determination
UPR Biomarkers in Acute Leukemias
Acute Myeloid Leukemia (AML)
Recent investigations have established the prognostic significance of UPR-related gene signatures in AML, a highly aggressive hematopoietic malignancy characterized by uncontrolled expansion of undifferentiated myeloid cells [133] [89]. Researchers have developed multi-gene UPR signatures that effectively stratify AML patients into distinct molecular subtypes with significant differences in clinical outcomes and treatment responses.
UPR-Related Molecular Subtypes in AML: Through consensus clustering of URGs, AML patients can be classified into two distinct subtypes (Cluster A and Cluster B) with markedly different survival outcomes [133] [89]. Cluster B patients demonstrate significantly poorer prognosis, elevated stromal/immune microenvironment scores, increased immune cell infiltration, and higher expression of immune checkpoint molecules, suggesting heightened sensitivity to immunotherapy approaches [133].
Six-Gene URGsig for Prognostic Stratification: A robust UPR-related gene signature (URGsig) comprising six genes (DNAJC10, DNAJB11, ANXA11, MBTPS1, SOD1, and VCP) has been constructed and validated [133] [89]. This signature stratifies AML patients into high-risk and low-risk subgroups with dramatically different overall survival. High-risk patients exhibit increased mortality, shorter survival times, distinct mutation profiles (higher FLT3, NPM1, and DNMT3A mutations), and enhanced immunosuppressive characteristics including M2 macrophage polarization and immune checkpoint expression [133].
Table 1: UPR-Related Gene Signatures in Acute Myeloid Leukemia
| Gene Signature | Component Genes | Prognostic Value | Biological Implications | Clinical Applications |
|---|---|---|---|---|
| 6-Gene URGsig [133] [89] | DNAJC10, DNAJB11, ANXA11, MBTPS1, SOD1, VCP | High-risk group: poorer overall survival (P<0.001) | High-risk: immunosuppressive TME, M2 macrophage polarization, immune checkpoint expression | Prognostic stratification, immunotherapy response prediction |
| UPR Molecular Subtypes [133] | 22 prognostic URGs | Cluster B: worse prognosis than Cluster A | Cluster B: elevated immune infiltration, checkpoint expression | Patient stratification for targeted immunotherapies |
| 4-Gene Hepatocellular Carcinoma Signature [134] | ATF4, GOSR2, PDIA6, SRPRB | High-risk: worse clinical prognosis | High-risk: immunosuppressive phenotype, immune escape | Sorafenib sensitivity prediction |
Functional Validation of UPR Biomarkers
The clinical relevance of URGsig components has been verified through multiple experimental approaches. Quantitative PCR analysis of AML patient bone marrow samples demonstrated significantly elevated expression of DNAJC10, VCP, and ANXA11 compared to control samples, with relapse patients showing further increased DNAJC10 expression and decreased DNAJB11 expression [133]. These findings validate the bioinformatics predictions and strengthen the utility of these markers for disease monitoring.
URGsig not only predicts prognosis but also informs therapeutic strategies. High-risk AML patients display lower sensitivity to conventional chemotherapy agents but increased responsiveness to sorafenib and immunotherapies, particularly anti-CTLA4 treatment [133]. This suggests UPR activation creates a therapeutic vulnerability that can be exploited with targeted approaches.
UPR Biomarkers in Solid Tumors
Hepatocellular Carcinoma (HCC)
In hepatocellular carcinoma, UPR-related genes show distinctive expression patterns with significant prognostic implications. Most URGs are upregulated in HCC samples compared to normal tissue, enabling molecular subtyping based on UPR activation status [134]. Cluster 1 patients exhibit higher UPR gene expression, poorer prognosis, and increased expression of immunosuppressive factors compared to Cluster 2, suggesting greater propensity for immune evasion during immunotherapy [134].
A validated 4-gene UPR signature (ATF4, GOSR2, PDIA6, SRPRB) effectively stratifies HCC patients by risk, with high-risk patients demonstrating worse clinical outcomes and differential responses to sorafenib treatment [134]. Functional studies reveal that sorafenib treatment modulates this signature, upregulating ATF4 while downregulating GOSR2, PDIA6, and SRPRB in HCC cells, providing mechanistic insights into treatment response and resistance [134].
Sarcomas and Bone Tumors
Sarcomas represent a heterogeneous group of malignant tumors with demonstrated UPR activation correlating with disease aggressiveness and outcomes [123] [4]. In osteosarcoma, the most extensively studied bone sarcoma subtype, UPR markers show upregulated expression associated with poorer prognosis [123]. UPR activation contributes to key aspects of sarcoma progression including proliferation, chemotherapy resistance, immune evasion, and angiogenesis [123].
Multiple myeloma, a hematologic malignancy with significant bone involvement, exhibits elevated ER stress proteins that correspond with adverse treatment outcomes [4]. The UPR enables myeloma cell survival within the bone marrow microenvironment, with specific branches supporting progression and treatment resistance.
Gynecologic Cancers
Comprehensive analyses across gynecologic malignancies (ovarian, endometrial, cervical) reveal consistent associations between UPR activation and aggressive phenotypes [132]. In epithelial ovarian cancer, GRP78, ATF6, and PERK overexpression correlates with inferior survival and chemoresistance, supporting their utility as biomarkers and therapeutic targets [132]. Endometrial cancer demonstrates UPR gene signature stratification of prognosis and immune infiltration, suggesting risk-adapted therapeutic strategies, while cervical cancer leverages PERK/IRE1 signaling for therapy tolerance and dormancy maintenance [132].
Table 2: UPR Biomarkers Across Solid Tumor Types
| Tumor Type | Key UPR Biomarkers | Prognostic Value | Therapeutic Implications |
|---|---|---|---|
| Hepatocellular Carcinoma [134] | ATF4, GOSR2, PDIA6, SRPRB | High expression correlates with poor prognosis | Sorafenib sensitivity prediction |
| Ovarian Cancer [132] | GRP78/HSPA5, ATF6, PERK | Overexpression associates with inferior survival and chemoresistance | Biomarkers for treatment selection and combination therapies |
| Sarcomas [123] | IRE1, PERK, ATF6 pathway components | Upregulation associated with aggressive disease | Potential predictors of response to UPR-targeted therapies |
| Gynecologic Cancers [132] | CHOP, XBP1, ATF4 | Activation correlates with advanced disease | Guidance for UPR modulator combinations with immunotherapy |
Experimental Methodologies for UPR Biomarker Analysis
Bioinformatics Pipelines for UPR Signature Development
The development and validation of UPR-related prognostic signatures follows a systematic bioinformatics workflow incorporating multiple computational approaches:
Data Acquisition and Preprocessing: Gene expression profiles and corresponding clinical data are obtained from public repositories including The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and International Cancer Genome Consortium (ICGC) [133] [89]. RNA-sequencing data typically undergoes transformation to transcripts per million (TPM) followed by log2 transformation to normalize expression distributions.
URG Selection and Molecular Subtyping: URGs are identified from databases such as GeneCards using keywords "unfolded protein response" with relevance score thresholds (typically >5) [133] [89]. Univariate Cox regression analysis identifies prognosis-associated URGs (p<0.01), followed by consensus clustering to define UPR-related molecular subtypes. Cluster stability is validated through iterative resampling (1000 repetitions) and t-distributed stochastic neighbor embedding (t-SNE) analysis [89].
Prognostic Model Construction: Least absolute shrinkage and selection operator (LASSO) Cox regression analysis selects significantly prognostic URGs, followed by multivariate Cox regression to identify independently prognostic genes for signature development [133] [89]. Risk scores are calculated using the formula: Risk score = Σ(Coef(Genei) × Exp(Genei)), with patients stratified into high- and low-risk groups based on median risk score.
Validation and Clinical Correlation: Signature performance is validated in independent cohorts with assessment of receiver operating characteristic (ROC) curves for predictive capability [89]. Subgroup analyses evaluate prognostic performance across clinical features including age, cytogenetic risk, and mutation status (FLT3, DNMT3A, TP53) [133].
Diagram Title: UPR Biomarker Research Workflow
Functional Characterization Approaches
Comprehensive bioinformatics analyses elucidate the biological implications of UPR signatures:
Differential Expression and Enrichment Analysis: Differentially expressed genes (DEGs) between risk subgroups are identified (|log2FC|≥1, P<0.05) followed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis [89]. Gene Set Enrichment Analysis (GSEA) reveals differentially enriched pathways between subgroups.
Tumor Microenvironment Evaluation: Immune cell infiltration patterns are assessed using CIBERSORT or similar deconvolution algorithms [133] [89]. ESTIMATE algorithm calculates immune, stromal, and microenvironment scores, while immune-related pathway activities are evaluated through principal component analysis.
Genomic Alteration Analysis: Somatic mutation and copy number variation (CNV) patterns are analyzed using tools like "maftools" [89]. Genomic Identification of Significant Targets in Cancer (GISTIC) algorithm identifies significant CNV regions, while protein-protein interaction networks are constructed using STRING database.
Therapeutic Response Prediction: Drug sensitivity is predicted using Genomics of Drug Sensitivity in Cancer (GDSC) database, while immunotherapy response is evaluated through immune checkpoint expression and tumor inflammation signatures [133] [89].
Table 3: Key Research Reagents and Resources for UPR Biomarker Studies
| Resource Category | Specific Tools/Reagents | Application/Function | Key Features |
|---|---|---|---|
| Bioinformatics Databases | TCGA-LAML, GEO datasets, ICGC | Source of gene expression and clinical data | Standardized processing, clinical annotation |
| Gene Reference | GeneCards | URG identification and annotation | Relevance scoring, functional information |
| Analysis Tools | CIBERSORT, ESTIMATE, XCell | Tumor microenvironment deconvolution | Immune cell infiltration quantification |
| Experimental Reagents | Tunicamycin, Thapsigargin | ER stress induction | N-linked glycosylation inhibition, calcium disruption |
| Therapeutic Inhibitors | STF-083010 (IRE1α RNase inhibitor), MKC-8866 | UPR pathway modulation | Mechanism validation, combination therapies |
| Validation Methods | qRT-PCR, Immunohistochemistry | Biomarker expression validation | Clinical sample verification |
Clinical Applications and Therapeutic Implications
The prognostic value of UPR signatures extends beyond survival prediction to informing therapeutic strategies across cancer types. Integration of URGsig with clinical variables (age, cytogenetic risk, mutation status) generates nomograms with demonstrated predictive accuracy for survival probability (AUC 0.912 for 5-year overall survival in AML) [133] [89].
UPR activation status correlates with differential responses to conventional and targeted therapies. High UPR activity generally associates with resistance to standard chemotherapy but increased sensitivity to specific targeted agents and immunotherapies [133] [132]. This has important implications for treatment selection, particularly in refractory or high-risk disease.
The UPR significantly influences the tumor immune microenvironment, with high-risk UPR profiles characterized by immunosuppressive features including M2 macrophage polarization, T-cell exhaustion, and enhanced immune checkpoint expression [133] [132]. These findings provide rationale for combining UPR-modulating agents with immunotherapy, particularly in UPR-activated, immunologically "cold" tumors.
UPR-related gene signatures represent robust biomarkers for diagnostic and prognostic applications across acute leukemias and solid tumors. The consistent demonstration that UPR activation correlates with aggressive disease features, therapy resistance, and altered immune microenvironments underscores the fundamental role of proteostatic stress responses in cancer pathogenesis. Future research directions should focus on validating UPR signatures in prospective clinical trials, defining context-specific vulnerabilities within UPR pathways, and developing biomarker-driven treatment strategies that combine UPR-targeted agents with conventional chemotherapy, targeted therapy, and immunotherapy. The integration of UPR biomarkers into clinical decision-making promises to enable more precise risk stratification and personalized therapeutic approaches for cancer patients.
Comparative Analysis of UPR Activation in Neurodegenerative Models (Alzheimer's, Parkinson's, ALS)
The endoplasmic reticulum (ER) serves as a critical organelle for protein synthesis, folding, and post-translational modifications in eukaryotic cells. Maintenance of ER proteostasis is essential for cellular function, particularly in neurons with high metabolic demands and secretory activity. When the protein-folding capacity of the ER is overwhelmed by accumulated misfolded or unfolded proteins, cells trigger an evolutionarily conserved adaptive mechanism known as the unfolded protein response (UPR) [3]. The UPR is initiated through three ER transmembrane sensors: protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) [3] [114]. Under normal conditions, these sensors are maintained in an inactive state through binding to the ER chaperone BiP/GRP78. During ER stress, BiP dissociates to bind misfolded proteins, activating these sensors and initiating downstream signaling cascades aimed at restoring proteostasis [114].
In neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), chronic ER stress and dysregulated UPR signaling contribute significantly to disease pathogenesis [135] [136]. The accumulation of disease-specific misfolded proteins—such as amyloid-β and tau in AD, α-synuclein in PD, and TDP-43 or mutant SOD1 in ALS—triggers persistent UPR activation that often transitions from an adaptive to a maladaptive, pro-apoptotic response [114]. This comparative analysis examines the specific patterns of UPR activation across these neurodegenerative models, highlighting both shared mechanisms and disease-specific alterations that present potential therapeutic targets for intervention.
UPR Signaling Pathways: Core Mechanisms
The Three Arms of the Unfolded Protein Response
The UPR comprises three distinct signaling branches initiated by PERK, IRE1, and ATF6, which work in concert to mitigate ER stress but can also trigger apoptosis under irremediable stress conditions [3].
PERK-eIF2α Pathway: Following its activation and dimerization, PERK phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α) at Ser51 [137] [114]. This phosphorylation event globally attenuates protein translation to reduce the protein-folding burden on the ER while simultaneously promoting the selective translation of specific mRNAs, including that of activating transcription factor 4 (ATF4) [137]. ATF4 then upregulates genes involved in antioxidant response, amino acid metabolism, and apoptosis. Under prolonged ER stress, ATF4 induces the expression of the pro-apoptotic transcription factor C/EBP homologous protein (CHOP), which promotes cell death by regulating downstream targets including Bcl-2 family proteins and death receptor 5 [138] [3].
IRE1-XBP1 Pathway: IRE1 possesses both kinase and endoribonuclease activities. Upon ER stress, IRE1 oligomerizes, autophosphorylates, and activates its RNase domain, which catalyzes the unconventional splicing of X-box binding protein 1 (XBP1) mRNA [138] [3]. The spliced XBP1 (XBP1s) functions as a potent transcription factor that upregulates genes encoding ER chaperones, components of ER-associated degradation (ERAD), and lipid biosynthesis enzymes to expand the ER's protein-folding capacity and facilitate the clearance of misfolded proteins [138]. Under severe or chronic stress, IRE1 can also initiate regulated IRE1-dependent decay (RIDD) of mRNAs and pro-apoptotic signaling through TRAF2 and JNK activation [3].
ATF6 Pathway: ATF6 is a type II ER transmembrane protein that translocates to the Golgi apparatus under stress conditions [3]. There, it undergoes proteolytic cleavage by site-1 protease (S1P) and site-2 protease (S2P), releasing its cytosolic domain (ATF6f). This active fragment functions as a transcription factor that migrates to the nucleus and induces the expression of ER chaperones (including BiP), XBP1, and components of the ERAD pathway [3] [114].
Integrated Stress Response and Mitochondrial UPR
The integrated stress response (ISR) represents a converging point for stress signaling from multiple cellular compartments. Four kinases—PERK, PKR, GCN2, and HRI—can phosphorylate eIF2α in response to diverse stressors such as ER stress, viral infection, amino acid deprivation, and heme deficiency [137]. This phosphorylation integrates these distinct stress signals into a common adaptive output, primarily mediated by ATF4. Furthermore, crosstalk exists between the UPR and the mitochondrial unfolded protein response (UPRmt), a compensatory mechanism activated upon mitochondrial proteostasis disruption [127]. Key transcription factors including ATF5, ATF4, and CHOP participate in both UPR and UPRmt, facilitating inter-organellar communication and coordinated stress adaptation [127].
Disease-Specific UPR Activation Patterns
Alzheimer's Disease
In Alzheimer's disease models, the accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau directly induces ER stress and activates all three branches of the UPR [114]. The PERK-eIF2α pathway is particularly prominent in AD pathogenesis. Phosphorylated PERK and eIF2α levels are elevated in the brains of AD patients and transgenic mouse models [114]. This sustained PERK activation leads to persistent translational repression and maladaptive ATF4/CHOP signaling, which contributes to synaptic dysfunction and neuronal apoptosis. The IRE1α-XBP1 pathway is also activated in AD, with increased XBP1 splicing observed in response to Aβ exposure. However, chronic IRE1 activation may engage RIDD and JNK-dependent apoptotic signaling, exacerbating neurodegeneration [114]. ATF6 activation has been demonstrated in AD models, where it initially provides a protective function by enhancing ER chaperone expression, though its beneficial effects may be overwhelmed as disease progresses [114].
Table 1: UPR Activation Markers in Alzheimer's Disease Models
| UPR Pathway | Key Activation Markers | Cellular Consequences | Therapeutic Implications |
|---|---|---|---|
| PERK | Increased p-PERK, p-eIF2α, ATF4, CHOP | Persistent translational repression, synaptic loss, neuronal apoptosis | PERK inhibitors (e.g., GSK2606414), ISRIB |
| IRE1 | Increased p-IRE1, XBP1 splicing, RIDD activity | Initial adaptive response, later pro-apoptotic signaling via JNK | IRE1 modulators (e.g., KIRA6) |
| ATF6 | Increased cleaved ATF6, upregulated BiP/GRP78 | Enhanced chaperone expression, increased ERAD capacity | ATF6 activators (e.g., AA147) |
Parkinson's Disease
Parkinson's disease models, characterized by the accumulation of misfolded α-synuclein and loss of dopaminergic neurons, demonstrate distinct UPR activation patterns [135]. The PERK pathway is significantly activated in both cellular and animal models expressing mutant or aggregated α-synuclein, with elevated levels of p-eIF2α and CHOP contributing to dopaminergic neuronal death [135]. The IRE1-XBP1 pathway shows a dual role in PD—XBP1 splicing has demonstrated protective effects in some models, while IRE1 hyperactivation promotes apoptosis through ASK1-JNK signaling and RIDD activity [135] [139]. ER stress in PD is not confined to the central nervous system but extends to peripheral tissues, including skeletal muscle, where it contributes to non-motor symptoms such as muscle wasting and weakness [135]. This systemic involvement highlights the potential for targeting UPR pathways therapeutically to address both central and peripheral pathology in PD.
Table 2: UPR Activation Markers in Parkinson's Disease Models
| UPR Pathway | Key Activation Markers | Cellular Consequences | Therapeutic Implications |
|---|---|---|---|
| PERK | Increased p-PERK, p-eIF2α, CHOP in substantia nigra | Dopaminergic neuron apoptosis, motor deficits | Chemical chaperones (e.g., TUDCA, PBA) |
| IRE1 | Variable XBP1 splicing, increased JNK phosphorylation | Context-dependent protective or pro-death outcomes | IRE1 kinase inhibitors |
| ATF6 | Activated ATF6, upregulated ER chaperones | Enhanced α-synuclein clearance, neuronal protection | Small molecule ATF6 activators |
Amyotrophic Lateral Sclerosis
In ALS models, including those expressing mutant SOD1, TDP-43, or FUS, UPR activation is particularly prominent in motor neurons and contributes to disease pathogenesis [135] [136]. The PERK-eIF2α pathway is robustly activated in ALS, with elevated p-eIF2α observed in both sporadic and familial ALS cases. However, the sustained activation of this pathway leads to maladaptive translational repression and CHOP-mediated apoptosis of vulnerable motor neurons [135]. The IRE1-XBP1 pathway is also activated in ALS, with evidence of increased XBP1 splicing in spinal cord samples. Interestingly, genetic studies have demonstrated that XBP1 deletion in the nervous system significantly ameliorates disease progression in SOD1 mutant mice, suggesting a detrimental role of chronic IRE1-XBP1 signaling in ALS [135]. ER stress in ALS extends beyond motor neurons to include skeletal muscle, where it contributes to muscle denervation, atrophy, and functional impairment [135].
Table 3: UPR Activation Markers in Amyotrophic Lateral Sclerosis Models
| UPR Pathway | Key Activation Markers | Cellular Consequences | Therapeutic Implications |
|---|---|---|---|
| PERK | Elevated p-PERK, p-eIF2α in motor neurons | Impaired protein synthesis, CHOP-mediated apoptosis | PERK inhibitors, GADD34 modulators |
| IRE1 | Increased XBP1 splicing, RIDD activation | Motor neuron death, muscle denervation | XBP1 genetic manipulation |
| ATF6 | Activated ATF6 fragment | Compensatory chaperone induction | Combination approaches |
Experimental Models and Methodologies
In Vitro Models and Assessment Techniques
Cell Culture Models: Neurodegenerative disease modeling utilizes various cell systems, including primary neuronal cultures, neuroblastoma cell lines (e.g., SH-SY5Y), and induced pluripotent stem cell (iPSC)-derived neurons. These models can be subjected to disease-relevant stressors such as Aβ oligomers for AD, pre-formed α-synuclein fibrils for PD, or expressed mutant proteins (SOD1, TDP-43) for ALS [114]. To specifically induce ER stress, researchers commonly use pharmacological agents including tunicamycin (N-glycosylation inhibitor), thapsigargin (SERCA pump inhibitor), and brefeldin A (protein transport inhibitor) [140].
UPR Activation Assessment: Western blotting is employed to detect protein levels and phosphorylation status of UPR sensors (p-PERK, p-IRE1, p-eIF2α) and effectors (ATF4, CHOP, XBP1s) [114]. Quantitative RT-PCR and RNA sequencing can monitor UPR-related gene expression changes, while specialized reporter constructs (e.g., XBP1 splicing reporters, ATF6 luciferase reporters) enable dynamic tracking of specific UPR branches in live cells [138] [114]. Immunofluorescence and immunohistochemistry allow spatial localization of UPR activation within subcellular compartments or specific cell populations in co-culture systems.
In Vivo Models and Translational Approaches
Transgenic Animal Models: AD research utilizes models such as APP/PS1 and 5xFAD mice; PD studies employ α-synuclein overexpression models (e.g., Thy1-αSyn, A53T transgenic mice) and toxin-based models (e.g., MPTP, 6-OHDA); ALS research commonly uses SOD1G93A and TDP-43 transgenic mice [114]. These models enable investigation of cell-type-specific UPR activation through techniques like immunohistochemistry on tissue sections and laser capture microdissection followed by transcriptomic analysis.
Therapeutic Intervention Studies: Preclinical studies test UPR-modifying compounds, including PERK inhibitors (GSK2606414), IRE1 modulators (KIRA6, KIRA7), and chemical chaperones (4-PBA, TUDCA) [114] [136]. These interventions are typically administered at various disease stages to assess their impact on UPR biomarkers, neuronal survival, and functional outcomes. Behavioral tests (e.g., rotarod, grip strength, cognitive assays) correlate UPR modulation with neurological function, while electrophysiological recordings assess synaptic integrity.
Visualization of UPR Signaling Pathways
UPR Signaling in Neurodegeneration
Research Reagent Solutions
Table 4: Essential Research Reagents for UPR Studies in Neurodegeneration
| Reagent Category | Specific Examples | Research Applications | Key Findings Enabled |
|---|---|---|---|
| ER Stress Inducers | Tunicamycin, Thapsigargin, Brefeldin A | Induction of controlled ER stress in cellular models | Mechanism of UPR activation and resolution |
| UPR Pathway Reporters | XBP1 splicing reporters, ATF6 luciferase reporters, CHOP-GFP constructs | Real-time monitoring of specific UPR branches | Kinetics and magnitude of pathway activation |
| UPR Modulators | PERK inhibitors (GSK2606414), IRE1 modulators (KIRA6), Chemical chaperones (TUDCA, 4-PBA) | Therapeutic targeting of specific UPR components | Pathway-specific functional outcomes |
| Antibodies for UPR Detection | Anti-p-PERK, Anti-p-eIF2α, Anti-CHOP, Anti-XBP1s, Anti-BiP/GRP78 | Immunodetection of UPR activation in cells and tissues | Quantitative assessment of UPR in disease models |
| Disease-Associated Protein Constructs | Mutant APP/PS1, A53T α-synuclein, SOD1G93A, TDP-43 mutants | Modeling proteinopathy-specific UPR activation | Disease-specific UPR signatures |
This comparative analysis reveals that while Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis share common features of UPR activation in response to protein misfolding, each neurodegenerative condition exhibits distinct patterns of ER stress pathway engagement. The PERK-eIF2α pathway emerges as a central player across all three diseases, though its temporal activation and functional consequences display disease-specific characteristics. The transition from adaptive to maladaptive UPR signaling represents a critical juncture in neurodegenerative pathogenesis, with persistent CHOP induction and compromised protein synthesis capacity contributing significantly to neuronal dysfunction and death.
The interconnectedness of cellular stress responses is highlighted by the crosstalk between UPR, mitochondrial quality control mechanisms, and the integrated stress response [127] [137]. Future therapeutic strategies should consider these complex interactions and the temporal aspects of UPR activation in neurodegeneration. The development of branch-specific UPR modulators that can enhance adaptive signaling while suppressing pro-apoptotic outputs holds promise for treating these devastating disorders. Furthermore, the extension of ER stress beyond the central nervous system to peripheral tissues, particularly skeletal muscle, suggests that therapeutic targeting of UPR pathways may address both central and systemic manifestations of neurodegenerative diseases [135].
The unfolded protein response (UPR), traditionally viewed as a cell-autonomous process, exhibits sophisticated cell-non-autonomous signaling capabilities that enable inter-tissue communication of proteostatic stress. This in-depth technical guide synthesizes current knowledge on the mechanisms and experimental validation of transcellular UPR signaling networks. We provide a comprehensive analysis of the molecular basis for UPR signal transmission, detailed methodological approaches for investigating these phenomena, and cutting-edge visualization of the complex signaling pathways involved. The emerging paradigm of cell-non-autonomous UPR has profound implications for understanding systemic regulation of proteostasis, organismal aging, and the progression of neurodegenerative diseases, offering novel therapeutic targets for modulating stress response pathways across tissues.
The endoplasmic reticulum (ER) serves as a crucial organelle for protein synthesis, folding, and quality control, with its proper function being essential for cellular homeostasis [1] [97]. When protein folding demand exceeds capacity, accumulating unfolded or misfolded proteins trigger ER stress, activating an adaptive mechanism known as the unfolded protein response (UPR) [3] [6]. The UPR is orchestrated by three primary ER transmembrane sensors: inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [3] [1]. Under normal conditions, these sensors are maintained in an inactive state through association with the chaperone BiP (binding immunoglobulin protein); however, upon ER stress, BiP dissociates to bind misfolded proteins, leading to sensor activation and initiation of downstream signaling cascades [3].
Historically, the UPR was considered exclusively cell-autonomous—activated within and acting upon individual stressed cells to restore proteostasis through mechanisms such as translational attenuation, chaperone upregulation, and ER-associated degradation (ERAD) [3] [1]. However, emerging evidence has fundamentally transformed this paradigm, revealing that UPR activation can be communicated between cells and tissues, operating in a cell-non-autonomous manner [141]. This inter-tissue communication allows localized stress experiences to elicit coordinated organism-wide responses, representing a sophisticated strategy for systemic stress adaptation.
This technical guide examines the molecular mechanisms, experimental validation methodologies, and implications of cell-non-autonomous UPR within the broader context of endoplasmic reticulum research. By integrating current findings and technical approaches, we aim to provide researchers with a comprehensive framework for investigating these transcellular signaling networks.
Molecular Mechanisms of Cell-Non-Autonomous UPR
Mitochondrial UPR as a Model for Transcellular Stress Signaling
The mitochondrial unfolded protein response (UPRmt) provides the most clearly established model for cell-non-autonomous stress signaling [141]. In Caenorhabditis elegans, mitochondrial stress in specific tissues triggers a retrograde signaling pathway that activates protective gene expression programs not only within the stressed tissue but also in distant, unstressed tissues [141]. This systemic response involves stress-induced secretion of mitokine molecules that communicate mitochondrial status throughout the organism.
Table 1: Key Features of Cell-Non-Autonomous UPRmt Signaling
| Feature | Mechanism | Experimental Evidence |
|---|---|---|
| Signal Origin | Mitochondrial proteostasis disruption | Tissue-specific mitochondrial complex knockdown |
| Signal Nature | Secreted peptides/hormones ("mitokines") | Neuroendocrine signaling components identified |
| Signal Transmission | Circulatory system/neuronal pathways | Demonstrated in worm, fly, and mouse models |
| Distant Tissue Response | Nuclear translocation of transcription factors (ATFS-1, ATF4, ATF5) | Activation of mitochondrial chaperone expression |
| Physiological Outcome | Enhanced stress resistance, metabolic adaptation, lifespan modulation | Longevity studies in C. elegans |
The UPRmt signaling cascade involves several key steps: (1) mitochondrial protein folding stress activates the transcription factor ATFS-1 in C. elegans (orthologs ATF4/ATF5 in mammals), (2) downstream signaling through neuroendocrine pathways, and (3) induction of mitochondrial chaperones and proteases in distant tissues [141]. This cell-non-autonomous activation enhances organismal stress resistance and influences metabolic homeostasis and lifespan [141].
ER UPR Cell-Non-Autonomous Signaling Pathways
While less comprehensively characterized than UPRmt, emerging evidence indicates that ER UPR also exhibits cell-non-autonomous characteristics. The signaling mechanisms potentially involve:
- Secreted Stress Ligands: ER-stressed cells may release signaling molecules (e.g., growth factors, cytokines, or specific peptides) that activate UPR pathways in recipient cells.
- Extracellular Vesicles: Stressed cells might package UPR-activated transcription factors or regulatory RNAs into exosomes or microvesicles for intercellular communication.
- Metabolite Signaling: ER stress alters cellular metabolism, potentially leading to secretion of metabolic byproducts that influence recipient cell proteostasis.
The IRE1-XBP1 branch appears particularly important in these communication events, potentially through regulated release of spliced XBP1 mRNA or XBP1s-dependent production of secreted factors [3]. Similarly, PERK-mediated regulation of the integrated stress response through eIF2α phosphorylation may influence secretion of proteins that modulate UPR in distant cells [3] [17].
Experimental Approaches for Validating Cell-Non-Autonomous UPR
Model Systems and Genetic Tools
Establishing appropriate model systems is fundamental for investigating cell-non-autonomous UPR phenomena. Multiple experimental approaches have proven effective:
Table 2: Model Systems for Cell-Non-Autonomous UPR Research
| Model System | Advantages | Key Genetic Tools | Limitations |
|---|---|---|---|
| C. elegans | Well-characterized neuroendocrine system, transparent for imaging, genetic tractability | Tissue-specific RNAi, UPR reporter strains (hsp-4::GFP, hsp-6::GFP), cell-specific ablation | Limited mammalian relevance, simple tissue organization |
| D. melanogaster | Complex organ systems, genetic manipulability, established stress paradigms | GAL4/UAS system for tissue-specific manipulation, fluorescent UPR reporters | Less developed biochemical tools compared to mammalian systems |
| Mus musculus | High physiological relevance to humans, complex tissue interactions | Cre-lox system for tissue-specific gene manipulation, UPR reporter mice | Technical complexity, higher costs, ethical considerations |
| Mammalian Cell Coculture | Precise control of experimental conditions, facile manipulation | Transwell systems, conditioned media transfer, CRISPR/Cas9 editing | May oversimplify in vivo complexity |
Methodological Framework for Experimental Validation
Establishing Tissue-Specific Stress Models
A. Genetic Approaches for Inducing Focal Stress:
- Implement tissue-specific CRISPR/Cas9 systems to knock out essential mitochondrial genes (e.g., components of electron transport chain) or ER quality control genes.
- Utilize tetracycline-inducible or tamoxifen-inducible systems for temporal control of stress induction in specific cell types.
- Employ photoactivatable stress inducers for precise spatial and temporal control of stress initiation.
B. Tissue-Specific UPR Reporter Systems:
- Generate transgenic animals expressing GFP under control of UPR-responsive promoters (e.g., BiP promoter for ER UPR, hsp-6/hsp-60 promoter for UPRmt) with tissue-specific Cre drivers.
- Implement multicolor reporter systems to simultaneously monitor different UPR branches in the same organism.
Demonstrating Signal Transmission
A. Parabiosis and Tissue Transplantation:
- Surgical joining of circulation between stressed and unstressed animals (parabiosis) to test for humoral transmission of UPR signals.
- Transplantation of stressed tissues into unstressed hosts to validate non-autonomous signaling.
B. Conditioned Media Transfer:
- Collect media from stressed cells and apply to unstressed reporter cells.
- Fractionate conditioned media to identify active signaling components.
- Treat conditioned media with proteases, nucleases, or specific inhibitors to characterize the nature of the transmitted signal.
Identifying Signaling Molecules
A. Secretome Analysis:
- Quantitative proteomics of conditioned media from stressed versus unstressed cells.
- RNA sequencing of extracellular vesicles from stressed cells.
- Metabolomic profiling to identify stress-induced secreted metabolites.
B. Functional Screening:
- siRNA screening targeting candidate secreted factors.
- Antibody neutralization of potential signaling molecules.
- Recombinant protein supplementation to test sufficiency for UPR induction.
Validation and Functional Assays
A. Confirmation of Non-Autonomous UPR Activation:
- Quantitative PCR for canonical UPR target genes (BiP, CHOP, XBP1s, mitochondrial chaperones) in distant tissues.
- Immunoblotting for UPR activation markers (phospho-eIF2α, XBP1s protein, ATF4/ATF5 accumulation).
- Immunohistochemistry to visualize activation in specific cell types within distant tissues.
B. Physiological Relevance Assessment:
- Stress resistance assays in recipient tissues.
- Metabolic profiling to identify systemic adaptations.
- Assessment of organismal lifespan or healthspan parameters.
Research Reagent Solutions for Cell-Non-Autonomous UPR Studies
Table 3: Essential Research Reagents for Cell-Non-Autonomous UPR Investigations
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| UPR Reporters | Hsp-4::GFP (C. elegans ER UPR), Hsp-6::GFP (C. elegans UPRmt), XBP1-splicing reporter, ATF6 luciferase reporter | Visualizing and quantifying UPR activation in specific tissues | Select reporters with demonstrated specificity; validate responsiveness to appropriate stressors |
| Stress Inducers | Tunicamycin (N-linked glycosylation inhibitor), Thapsigargin (SERCA inhibitor), DTT (reducing agent), Antimycin A (mitochondrial complex III inhibitor) | Experimental induction of organelle-specific stress | Titrate concentration to achieve sublethal stress; consider tissue-specific delivery methods |
| Genetic Tools | Tissue-specific Cre drivers, floxed UPR genes, RNAi constructs, CRISPR/Cas9 systems with tissue-specific promoters | Selective manipulation of UPR components in specific tissues | Verify recombination efficiency; control for off-target effects; use appropriate inducible systems |
| Signaling Inhibitors | ISRIB (integrated stress response inhibitor), KIRA6 (IRE1 inhibitor), GSK2606414 (PERK inhibitor) | Blocking specific UPR branches to test necessity | Assess specificity in your model system; monitor compensatory activation of alternative pathways |
| Secretome Analysis Tools | Extracellular vesicle isolation kits, proximity-dependent biotin identification (BioID), azidonorleucine labeling for nascent secreted proteins | Identifying candidate signaling molecules | Use appropriate controls for cellular contamination; consider temporal aspects of secretion |
Signaling Pathway Visualization
The following diagrams illustrate the core mechanisms of cell-non-autonomous UPR signaling, created using Graphviz DOT language with specified color palette and contrast requirements.
Cell-Non-Autonomous UPRmt Signaling Pathway
Diagram 1: Systemic UPRmt Signaling Cascade - This diagram illustrates the communication of mitochondrial stress from a stressed tissue to distant tissues via secreted factors (mitokines) traveling through circulation, leading to enhanced proteostasis in the recipient tissue.
Experimental Workflow for Validation
Diagram 2: Experimental Validation Workflow - This workflow outlines the three-phase approach for validating cell-non-autonomous UPR, progressing from model establishment through signal transmission confirmation to mechanistic elucidation.
Discussion and Future Perspectives
The emerging paradigm of cell-non-autonomous UPR represents a significant advancement in our understanding of how organisms coordinate proteostatic responses across tissues. The conserved nature of these signaling mechanisms from invertebrates to mammals [141] underscores their fundamental biological importance in maintaining organismal homeostasis under stress conditions.
Several critical questions remain unanswered and represent fertile ground for future investigation:
Molecular Identity of Signaling Factors: While the existence of transcellular UPR signals is well-established, the precise molecular identity of many of these signaling entities remains elusive. Systematic approaches to identify these factors will be essential for understanding the communication mechanisms.
Signal Specificity and Decoding: How do recipient cells distinguish between different types of organelle stress (ER vs. mitochondrial) and mount appropriate responses? The specificity mechanisms in cell-non-autonomous signaling require further elucidation.
Pathological Implications: Dysregulation of cell-non-autonomous UPR may contribute to disease progression, particularly in neurodegenerative contexts where protein aggregation spreads between connected brain regions [142] [6]. Understanding these mechanisms may reveal novel therapeutic approaches for conditions like Alzheimer's disease.
Therapeutic Manipulation: The ability to modulate cell-non-autonomous UPR signaling holds promise for treating proteostasis-related diseases. Potential strategies include enhancing protective signaling from healthy tissues to stressed ones or blocking maladaptive signaling that exacerbates pathology.
As research methodologies continue to advance, particularly in single-cell analysis and in vivo imaging, we anticipate rapid progress in deciphering the complex language of inter-tissue proteostatic communication. The integration of these findings with the broader context of organelle biology and organismal physiology will ultimately provide a more comprehensive understanding of how multicellular organisms maintain proteostasis in the face of constant environmental and metabolic challenges.
The endoplasmic reticulum (ER) is a critical organelle for protein folding, lipid synthesis, and calcium storage. The accumulation of unfolded or misfolded proteins within the ER lumen triggers a state known as ER stress, which activates a sophisticated adaptive mechanism termed the unfolded protein response (UPR) [3]. The UPR is orchestrated by three primary ER-resident transmembrane sensors: inositol-requiring enzyme 1 (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [2] [88]. Under normal conditions, these sensors are bound and inhibited by the chaperone protein GRP78 (BiP/HSPA5). An accumulation of unfolded proteins causes GRP78 to dissociate, leading to the activation of these sensors and the initiation of complex signaling pathways aimed at restoring proteostasis [88] [3].
In the context of cancer, tumor cells exploit the pro-survival facets of the UPR to thrive in the hostile tumor microenvironment, which is characterized by hypoxia, nutrient deprivation, and oxidative stress [17]. This reliance creates a therapeutic window, where targeted disruption of specific UPR branches can selectively impair cancer cell adaptation, evade apoptosis, and reverse therapeutic resistance [17] [123]. This guide provides a comprehensive technical overview of the current strategies for therapeutically validating UPR-targeted agents in vivo, detailing their efficacy, associated challenges, and essential experimental methodologies.
UPR Signaling Pathways and Their Therapeutic Exploitation
The three branches of the UPR employ distinct mechanisms to regulate cell fate. The diagram below illustrates the core signaling pathways and the points of therapeutic intervention.
Diagram 1: UPR Signaling Pathways and Therapeutic Inhibition. The graphic depicts the three primary UPR branches (IRE1α, PERK, ATF6) activated by ER stress, their downstream signaling leading to adaptive or apoptotic outcomes, and the points of intervention for key small-molecule inhibitors.
- The IRE1α Axis: IRE1α, the most evolutionarily conserved branch, possesses both kinase and endoribonuclease activities. Upon activation, it splices the mRNA of X-box binding protein 1 (XBP1), generating a potent transcription factor (XBP1s) that drives the expression of genes involved in ER chaperoning, lipid biosynthesis, and ER-associated degradation (ERAD) [143] [123]. Under severe or prolonged stress, IRE1α can also trigger apoptosis through Regulated IRE1-Dependent Decay (RIDD) of mRNAs and by activating the TRAF2/ASK1/JNK pathway [88] [123].
- The PERK Axis: PERK activation leads to the phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α), which transiently inhibits global protein translation to reduce the ER's protein load. However, this phosphorylation selectively promotes the translation of key transcripts, notably activating transcription factor 4 (ATF4). ATF4 upregulates genes involved in amino acid metabolism, antioxidant response, and, under chronic stress, the pro-apoptotic factor C/EBP-homologous protein (CHOP) [17] [3].
- The ATF6 Axis: ER stress triggers the translocation of ATF6 to the Golgi apparatus, where it is cleaved by proteases (S1P and S2P). The released cytosolic fragment (ATF6f) acts as a transcription factor, inducing genes encoding ER chaperones and components of the ERAD pathway, thereby enhancing the ER's folding and degradation capacity [88] [123].
Portfolio of UPR-Targeted Agents and In Vivo Efficacy
The therapeutic strategy of targeting the UPR involves either inhibiting pro-survival signals to induce proteotoxic cell death or hyperactivating the UPR beyond a manageable threshold to trigger apoptosis. The table below summarizes key UPR-targeted agents and their documented in vivo efficacy.
Table 1: UPR-Targeted Agents and Documented In Vivo Efficacy
| Target | Agent Name | Mechanism of Action | In Vivo Model(s) | Reported Efficacy & Key Findings |
|---|---|---|---|---|
| IRE1α RNase | B-I09 | Inhibits IRE1α's RNase activity, blocking XBP1 splicing [88]. | Multiple Myeloma models [88]. | Reduced tumor growth; synergistic with proteasome inhibitors [88]. |
| IRE1α RNase | MKC-8866 | IRE1α RNase inhibitor [88]. | Preclinical cancer models [88]. | Showed anti-tumor activity in preclinical studies [88]. |
| PERK | GSK2606414 | Potent and selective PERK kinase inhibitor [17] [88]. | Multiple xenograft models (e.g., glioblastoma) [17]. | Suppressed tumor growth; induced apoptosis. Notable on-target toxicity (weight loss, hyperglycemia) [17] [88]. |
| p97/VCP | CB-5083 | Inhibits p97 ATPase, disrupting ERAD and inducing UPR [144]. | Rhabdomyosarcoma (RMS) PDX models [144]. | Activated UPR (PERK-eIF2α-CHOP), induced apoptosis; superior bioavailability vs. HSP70 inhibitors [144]. |
| GRP78 | PAT-SM6 | Fully human IgM monoclonal antibody targeting cell-surface GRP78 [17]. | Preclinical cancer models [17]. | Induces immune-mediated cytotoxicity; inhibits pro-survival activity [17]. |
| GRP78 | HA15 | Small molecule binding GRP78's substrate-binding domain [17]. | Melanoma, breast cancer models [17]. | Inhibits chaperone/ATPase activity; induces ER stress and apoptosis [17]. |
| ATF6 | Ceapins | Specific inhibitors that block ATF6 transcriptional activity [17] [88]. | Preclinical studies [17] [88]. | Block ATF6α signaling without compensatory IRE1/XBP1 activation [17] [88]. |
Beyond single-agent efficacy, rational combination therapies are a major focus. For instance, in rhabdomyosarcoma, p97 inhibition activates the PERK-CHOP axis, and resistant tumors show elevated autophagy, nominating co-targeting of compensatory pathways as a promising strategy [144]. Combining UPR inhibitors with conventional chemotherapy, PARP inhibitors, or immunotherapy is also under active investigation to overcome treatment resistance [88] [99].
Experimental Workflow for In Vivo Therapeutic Validation
Robust in vivo validation is paramount for translating UPR-targeting agents. The following diagram and protocol detail a standard workflow for assessing the efficacy and mechanisms of these compounds.
Diagram 2: In Vivo Therapeutic Validation Workflow. A sequential pipeline for evaluating UPR-targeted agents from initial model setup to final mechanistic analysis.
Detailed Experimental Protocol
Step 1: Model Selection and Tumor Establishment
- Model Choice: Select patient-derived xenografts (PDXs) or cell line-derived xenografts (CDXs) based on genetic background and documented UPR dependency. For example, RMS PDX models were used to validate p97 inhibition [144], while gynecologic cancer models are relevant for IRE1/XBP1 inhibitors [88].
- Engraftment: Implant cancer cells/sub fragments subcutaneously into immunocompromised mice (e.g., NSG or nude mice). For studies involving the tumor-immune interface, humanized mouse models are increasingly required [17] [88].
Step 2: Treatment Arms and Dosing Regimen
- Arm Design: Include vehicle control, monotherapy arms (UPR inhibitor), standard-of-care chemotherapy, and combination arms.
- Dosing: Begin treatment when tumors reach a predefined volume (e.g., 100-150 mm³). Administer the UPR inhibitor at its established maximum tolerated dose (MTD) or biologically effective dose. Example: CB-5083 was administered orally in RMS models [144].
Step 3: In Vivo Efficacy Monitoring
- Tumor Volume: Measure tumor dimensions 2-3 times weekly using calipers. Calculate volume with the formula: (Length × Width²) / 2.
- Body Weight: Monitor mouse weight as a primary indicator of treatment-related toxicity.
- Advanced Imaging: Utilize modalities like ultrasound or bioluminescence imaging for deep-seated or metastatic tumors.
Step 4: Endpoint Tissue Collection
- At the study endpoint, harvest tumors and rapidly dissect them into sections for:
- Snap-freezing in liquid nitrogen for protein/RNA extraction.
- Formalin-fixation and paraffin-embedding (FFPE) for immunohistochemistry (IHC) and histology.
- Preservation in RNAlater for transcriptomic analyses.
Step 5: Ex Vivo Biomarker Analysis
- Western Blotting: Quantify key UPR protein markers from tumor lysates, including:
- IRE1α pathway: XBP1s protein, phospho-IRE1α.
- PERK pathway: Phospho-PERK, phospho-eIF2α, ATF4, CHOP.
- ATF6 pathway: Cleaved ATF6f.
- Apoptosis: Cleaved caspase-3, PARP cleavage [144].
- Quantitative PCR (qPCR): Measure transcript levels of UPR target genes (e.g., XBP1s, BiP, CHOP). Spliced XBP1 is a definitive marker of IRE1α activation [144].
- Immunohistochemistry (IHC): Spatial assessment of biomarker expression (e.g., CHOP, GRP78) and immune cell infiltration (e.g., CD8+ T cells) within the tumor microenvironment [88].
Step 6: Mechanism Deconvolution and Resistance Studies
- RNA Sequencing: Perform transcriptomic profiling on treated vs. control tumors to identify differentially expressed pathways, novel biomarkers, and potential resistance mechanisms (e.g., elevated autophagy genes as seen in p97 inhibitor-resistant RMS) [144].
- Combination Studies: Based on mechanistic insights, design subsequent in vivo studies to test rational combinations, such as adding an autophagy inhibitor to a UPR-targeted agent [144] [99].
The Scientist's Toolkit: Key Reagents for UPR Research
Table 2: Essential Research Reagents for Investigating the UPR
| Reagent Category | Specific Example(s) | Function & Application in UPR Research |
|---|---|---|
| ER Stress Inducers | Thapsigargin (Tg), Tunicamycin (Tm), Brefeldin A | Tool compounds used in vitro to induce ER stress and activate the UPR; essential for control experiments and pathway stimulation [2] [145]. |
| Small Molecule Inhibitors | GSK2606414 (PERKi), B-I09/MKC-8866 (IRE1α RNase i), CB-5083 (p97/VCP i), Ceapins (ATF6 i) | Pharmacologic tools to selectively inhibit specific UPR branches and assess functional outcomes in vitro and in vivo [17] [88] [144]. |
| Antibodies for Immunoblotting | Anti-phospho-PERK, Anti-phospho-eIF2α, Anti-ATF4, Anti-CHOP, Anti-XBP1s, Anti-cleaved Caspase-3 | Critical for detecting UPR activation and downstream apoptotic signaling in cell and tumor lysates via Western blot [144]. |
| qPCR Assays | Assays for XBP1 splicing, HSPA5 (BiP), DDIT3 (CHOP) | Gold-standard method for quantifying the splicing of XBP1 mRNA and measuring the transcriptional output of the UPR [144]. |
| Cell Viability Assays | CellTiter-Glo, MTT, Annexin V/PI staining | To quantify the cytotoxic and pro-apoptotic effects of UPR-modulating agents [144]. |
Challenges and Future Directions in UPR-Targeted Therapy
Despite the promising preclinical data, several significant challenges impede the clinical translation of UPR-targeted agents.
- On-Target Toxicity: The UPR is crucial for the function of normal secretory cells. PERK inhibition, for example, has been associated with pancreatic toxicity and weight loss in animal models, highlighting the potential for serious side effects [17] [88]. Developing optimal therapeutic windows and dosing schedules is critical.
- Context-Dependent Roles: The UPR is not universally pro-tumorigenic. In some contexts, certain UPR components can have tumor-suppressive functions. For instance, low XBP1 mRNA levels paradoxically predict poor outcomes in serous ovarian cancer [88]. This underscores the necessity for patient stratification and biomarker development to identify tumors that are truly "UPR-addicted" [17] [123].
- Tumor Heterogeneity and Adaptive Resistance: Tumors exhibit remarkable plasticity. Inhibition of one UPR branch can lead to the compensatory upregulation of another. Furthermore, as demonstrated with p97 inhibition, tumors can activate parallel survival pathways like autophagy to evade therapy [144] [99]. This necessitates combinatorial targeting of the UPR and its escape mechanisms.
- The Tumor-Immune Interface: The UPR is active not only in cancer cells but also in immune cells within the tumor microenvironment. IRE1α-XBP1 signaling in dendritic cells and macrophages can promote an immunosuppressive environment [17] [2]. This presents both a challenge and an opportunity, as combining UPR inhibitors with immunotherapy (e.g., anti-PD-1) is a promising strategy to enhance anti-tumor immunity [17] [88].
Future work must prioritize the development of more selective inhibitors, the identification of robust predictive biomarkers, and the design of sophisticated clinical trials that rationally combine UPR-targeted agents with other modalities to overcome resistance and improve patient outcomes.
Conclusion
The Unfolded Protein Response represents a fundamental cellular process with far-reaching implications in health and disease. This synthesis underscores the UPR's duality as both a protective mechanism and a contributor to pathology in cancer, neurodegeneration, and metabolic disorders. The experimental and therapeutic landscape is rapidly evolving, with promising small-molecule inhibitors moving into preclinical and clinical development. However, significant challenges remain, including the context-dependent outcomes of UPR modulation and the need for precise, cell-type-specific targeting strategies. Future research must focus on deciphering the intricate crosstalk between UPR branches and other cellular pathways, developing more sophisticated biomarkers for patient stratification, and designing combination therapies that leverage UPR inhibition or hyperactivation to overcome treatment resistance. A deepened understanding of UPR mechanics and its systemic effects will undoubtedly unlock novel, transformative approaches for a wide spectrum of human diseases.
References
- 1. Endoplasmic reticulum stress signalling - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC7379631/]
- 2. Endoplasmic reticulum stress and unfolded protein response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592176/]
- 3. Endoplasmic reticulum stress: molecular mechanism and ... [https://www.nature.com/articles/s41392-023-01570-w]
- 4. The role of the unfolded protein response pathway in bone ... [https://www.nature.com/articles/s41413-025-00457-6]
- 5. Targeting the Unfolded Protein Response with Natural ... [https://www.mdpi.com/1422-0067/26/18/8814]
- 6. Endoplasmic Reticulum Stress Coping Mechanisms and ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2019.00084/full]
- 7. ER stress as a sentinel mechanism for ER Ca2+ homeostasis [https://www.sciencedirect.com/science/article/pii/S0143416024001192]
- 8. Adaptive ER stress promotes mitochondrial remodelling ... [https://www.nature.com/articles/s41418-025-01603-7]
- 9. Unique and Conserved Endoplasmic Reticulum Stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12524277/]
- 10. Mechanism, regulation and functions of the unfolded protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8867924/]
- 11. Structure and Molecular Mechanism of ER Stress Signaling ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2019.00011/full]
- 12. Signaling Pathways from the Endoplasmic Reticulum and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3924831/]
- 13. Dynamic changes in complexes of IRE1α, PERK, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5994896/]
- 14. The Role of ER Stress and the Unfolded Protein Response ... [https://cgp.iiarjournals.org/content/22/3/363]
- 15. Feedback-responsive cell factories for dynamic modulation ... [https://www.nature.com/articles/s41467-025-58994-x]
- 16. IRE1 signaling increases PERK expression during chronic ... [https://www.nature.com/articles/s41419-024-06663-0]
- 17. Review Targeting the unfolded protein response in cancer [https://www.sciencedirect.com/science/article/abs/pii/S0006295225004861]
- 18. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5008685/]
- 19. Endoplasmic reticulum stress and unfolded protein ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1694102/full]
- 20. Controlling the unfolded protein response-mediated life ... [https://www.sciencedirect.com/science/article/abs/pii/S1044579X15000188]
- 21. The PERK-eIF2α-ATF4 Axis Is Involved in Mediating ER ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11939615/]
- 22. Activation of PERK/eIF2α/ATF4 signaling inhibits ERα ... [https://www.sciencedirect.com/science/article/pii/S1476558625000442]
- 23. Integrated stress response plasticity governs normal cell ... [https://www.nature.com/articles/s41418-024-01378-3]
- 24. The functions of IRE1α in neurodegenerative diseases [https://www.sciencedirect.com/science/article/abs/pii/S1568163722002161]
- 25. Review Getting RIDD of RNA: IRE1 in cell fate regulation [https://www.sciencedirect.com/science/article/abs/pii/S0968000414000334]
- 26. Regulated IRE1α-dependent decay (RIDD)-mediated ... [https://www.nature.com/articles/s41467-022-30159-0]
- 27. Decoding non-canonical mRNA decay by the endoplasmic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8674358/]
- 28. The IRE1α-XBP1 Pathway of the Unfolded Protein Response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2963107/]
- 29. Novel mechanism of enhancing IRE1α-XBP1 signalling via ... [https://www.nature.com/articles/srep24217]
- 30. IRE1α-XBP1 pathway promotes melanoma progression by ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-017-1147-2]
- 31. ATF6 Activated by Proteolysis Binds in the Presence of NF- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC86199/]
- 32. The Unfolded Protein Response and Autophagy as Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7550790/]
- 33. ATF6 is a transcription factor specializing in the regulation ... [https://pubmed.ncbi.nlm.nih.gov/18360008/]
- 34. ER stress signaling by regulated proteolysis of ATF6 [https://www.sciencedirect.com/science/article/abs/pii/S1046202304002373]
- 35. ER stress signaling by regulated proteolysis of ATF6 [https://pubmed.ncbi.nlm.nih.gov/15804611/]
- 36. Expression of UPR effector proteins ATF6 and XBP1 ... [https://www.nature.com/articles/s41419-019-1729-4]
- 37. Mutations in unfolded protein response regulator ATF6 ... [https://www.jci.org/articles/view/175562]
- 38. Dysregulation of Retinal and Photoreceptor Structural ... [https://pubmed.ncbi.nlm.nih.gov/39930229/]
- 39. Regulation of Apoptosis by the Unfolded Protein Response [https://pmc.ncbi.nlm.nih.gov/articles/PMC3684430/]
- 40. Mediators of endoplasmic reticulum stress-induced apoptosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC1559676/]
- 41. The balance between adaptive and apoptotic unfolded ... [https://www.sciencedirect.com/science/article/abs/pii/S0303720715300046]
- 42. ER Stress and the Unfolded Protein Response [https://pmc.ncbi.nlm.nih.gov/articles/PMC9735958/]
- 43. Signaling and induction of chaperone-mediated autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6103395/]
- 44. Regulation of autophagy by canonical and non ... [https://www.sciencedirect.com/science/article/abs/pii/S1044579X19303943]
- 45. Emerging mechanisms of the unfolded protein response in ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-023-01438-0]
- 46. sUPRa is a dual-color reporter for unbiased quantification ... [https://www.nature.com/articles/s41598-024-65611-2]
- 47. Endoplasmic reticulum stress and unfolded protein ... [https://pubmed.ncbi.nlm.nih.gov/41208954/]
- 48. Crosstalk Between Hypoxia and ER Stress Response [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.02951/full]
- 49. Physiological functions of endoplasmic reticulum stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3889286/]
- 50. Hypoxia, endoplasmic reticulum stress and chemoresistance [https://pubmed.ncbi.nlm.nih.gov/33423689/]
- 51. Endoplasmic Reticulum (ER) Stress and Hypoxia ... [https://www.sciencedirect.com/science/article/pii/S0021925819747280]
- 52. A guide to understanding endoplasmic reticulum stress in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7887651/]
- 53. Pharmacological landscape of endoplasmic reticulum stress [https://www.sciencedirect.com/science/article/abs/pii/S0014299925002638]
- 54. Frontiers | An unfolded protein response ( UPR )-signature regulated by... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1542650/full]
- 55. Decoding contextual crosstalk: revealing distinct interactions between... [https://cancerci.biomedcentral.com/articles/10.1186/s12935-024-03296-3]
- 56. A multi-omics analysis reveals the unfolded protein ... [https://www.nature.com/articles/s41467-020-16747-y]
- 57. JCI - Orchestrating the unfolded protein response in health and disease [https://www.jci.org/articles/view/16886]
- 58. Methods for Monitoring Endoplasmic Reticulum Stress and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2821749/]
- 59. Methods for studying ER stress and UPR markers in ... [https://pubmed.ncbi.nlm.nih.gov/25804744/]
- 60. Circulating protein biomarkers identified in two ... [https://www.nature.com/articles/s41598-025-23758-6]
- 61. Large-scale serum protein biomarkers discovery ... [https://www.nature.com/articles/s41467-025-64146-y]
- 62. Identification and validation of endoplasmic reticulum stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11959167/]
- 63. Protein kinase RNA- like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)- mediated endoplasmic reticulum stress- induced apoptosis in diabetic cardiomyopathy - Cardiovascular Diabetology [https://cardiab.biomedcentral.com/articles/10.1186/1475-2840-12-158]
- 64. The C/EBP Homologous Protein (CHOP) Transcription ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.03083/full]
- 65. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in ... [https://pubmed.ncbi.nlm.nih.gov/11779464/]
- 66. IRE1-mediated unconventional mRNA splicing and S2P ... [https://pubmed.ncbi.nlm.nih.gov/11850408/]
- 67. A novel role of ER stress signal transducer ATF6 in regulating ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-018-0412-x]
- 68. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress - PubMed [https://pubmed.ncbi.nlm.nih.gov/27211800/]
- 69. CHOP-independent apoptosis and pathway-selective ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2830287/]
- 70. XBP1 splicing contributes to endoplasmic reticulum stress ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11237843/]
- 71. Glucose sensing and the unfolded protein response [https://pubmed.ncbi.nlm.nih.gov/40272086/]
- 72. GlcNAc-1-P-transferase-tunicamycin complex structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5840018/]
- 73. Tunicamycin | Other Transferases [https://www.tocris.com/products/tunicamycin_3516]
- 74. Relationship of the structure and biological activity ... [https://www.sciencedirect.com/science/article/pii/S0021925819810803]
- 75. Thapsigargin, Origin, Chemistry, Structure-Activity ... [https://pubmed.ncbi.nlm.nih.gov/26429715/]
- 76. Thapsigargin [https://en.wikipedia.org/wiki/Thapsigargin]
- 77. Dithiothreitol [https://en.wikipedia.org/wiki/Dithiothreitol]
- 78. Endoplasmic reticulum stress and therapeutic strategies in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10956371/]
- 79. Emerging mechanisms of the unfolded protein response in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10832166/]
- 80. The Hidden Hand of Endoplasmic Reticulum Stress in ... [https://www.sciencedirect.com/science/article/abs/pii/S1040842825003865]
- 81. Small molecule strategies to harness the unfolded protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7667976/]
- 82. Endoplasmic Reticulum Stress in Hepatocellular Carcinoma [https://www.genesispub.org/endoplasmic-reticulum-stress-in-hepatocellular-carcinoma]
- 83. Endoplasmic Reticulum Stress and Unfolded Protein ... [https://pubmed.ncbi.nlm.nih.gov/40641847/]
- 84. Targeted inhibition of protein synthesis renders cancer ... [https://www.nature.com/articles/s41419-023-06055-w]
- 85. Endoplasmic Reticulum Stress in Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12179417/]
- 86. Endoplasmic reticulum stress—a key guardian in cancer [https://www.nature.com/articles/s41420-024-02110-3]
- 87. Modulation of Endoplasmic Reticulum Stress in ... [https://pubmed.ncbi.nlm.nih.gov/40650182/]
- 88. The Role of Endoplasmic Reticulum Stress and Unfolded ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12517228/]
- 89. Identification of a novel unfolded protein response related ... [https://www.nature.com/articles/s41598-025-91524-9]
- 90. PMV Pharma Presents Promising Phase 2 Trial Results for ... [https://www.quiverquant.com/news/PMV+Pharma+Presents+Promising+Phase+2+Trial+Results+for+Rezatapopt+at+2025+AACR-NCI-EORTC+Conference]
- 91. Targeting the Unfolded Protein Response with Natural Products [https://pmc.ncbi.nlm.nih.gov/articles/PMC12469893/]
- 92. 未折叠蛋白反应(UPR)及其在神经退行性疾病中的双重作用。 [https://suppr.wilddata.cn/browses/detail/pubmed/37817014]
- 93. Frontiers | Inhibition of pro-apoptotic UPR pathways PERK/CHOP and... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2025.1700897/abstract]
- 94. 陈畅课题组揭示枸杞水提物通过诱导线粒体UPR抑制蛋白聚集 [http://ibp.cas.cn/zyxw/2022tpxw/202411/t20241104_7420252.html]
- 95. 研究人员开发新策略为神经退行疾病治疗开新路 - 新闻 [https://news.sciencenet.cn/htmlnews/2025/9/551549.shtm]
- 96. Deep Visual Proteomics maps proteotoxicity in a genetic liver disease [https://www.nature.com/articles/s41586-025-08885-4?error=cookies_not_supported&code=24eddbbc-1f68-4469-8b41-6c97e446a9ac]
- 97. Endoplasmic reticulum stress: A key player in immune cell ... [https://www.sciencedirect.com/science/article/pii/S1044532325000260]
- 98. Gliadin-dependent UPR induction directly triggers the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12007252/]
- 99. Targeting proteostasis for cancer therapy: current advances ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02472-x]
- 100. The unfolded protein response and endoplasmic reticulum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6336407/]
- 101. The unfolded protein response supports cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3251055/]
- 102. Targeting endoplasmic reticulum stress: an innovative ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06076-3]
- 103. Unfolded protein response (UPR) integrated signaling ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-020-00212-1]
- 104. Phosphorylation at Ser724 of the ER stress sensor IRE1α ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9144033/]
- 105. Western blot protocol for low abundance proteins [https://www.abcam.com/en-us/technical-resources/protocols/western-blot-protocol-for-low-abundance-proteins]
- 106. Detecting Low Abundance Proteins in Western Blotting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/detecting-low-abundance-proteins-in-western-blotting.html]
- 107. Five Tips to Successful Western Blot of phospho-IRE1 ... [https://www.novusbio.com/node/12415896?srsltid=AfmBOopYNFn1A3bAIwsSxV4i8opaJ6Q-hkkU3sRqjKBUEu0_iizh4aHc]
- 108. A Guide to Modern Quantitative Fluorescent Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4354265/]
- 109. Phospho-IRE1 alpha (Ser724) Polyclonal Antibody (PA5- ... [https://www.thermofisher.com/antibody/product/Phospho-IRE1-alpha-Ser724-Antibody-Polyclonal/PA5-85738]
- 110. Understanding the Unfolded Protein Response (UPR) Pathway [https://pmc.ncbi.nlm.nih.gov/articles/PMC10902702/]
- 111. Measuring ER stress and the unfolded protein response using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3701721/]
- 112. The unfolded protein response mediates adaptation to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3057411/]
- 113. Table 1 Functions of the ER stress-induced genes considered ... [https://bmcvetres.biomedcentral.com/articles/10.1186/1746-6148-10-46/tables/1]
- 114. ER stress and UPR in Alzheimer's disease [https://www.nature.com/articles/s41419-022-05153-5]
- 115. Endoplasmic reticulum stress in cardiomyopathies [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2025.1577186/full]
- 116. Detecting and Quantitating Physiological Endoplasmic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3374842/]
- 117. Deciphering ER stress-unfolded protein response ... [https://www.sciencedirect.com/science/article/pii/S2211124724006867]
- 118. and therapeutic strategies in metabolic... Endoplasmic reticulum stress [https://molmed.biomedcentral.com/articles/10.1186/s10020-024-00808-9]
- 119. Dissecting Branch-Specific Unfolded Protein Response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12362115/]
- 120. Unfolded protein response - Wikipedia [https://en.wikipedia.org/wiki/Unfolded_protein_response]
- 121. Frontiers | ER and the Stress in Shaping Intestinal Tissue... UPR [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.02825/full]
- 122. Imaging of single cell responses to ER indicates that the... stress [https://pubmed.ncbi.nlm.nih.gov/25633195/]
- 123. The Unfolded Protein Response in Sarcomas - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606766/]
- 124. ONC201 induces the unfolded protein response (UPR) in high [https://pmc.ncbi.nlm.nih.gov/articles/PMC8124100/]
- 125. Termination of the unfolded protein response is guided by ... [https://www.nature.com/articles/s41467-022-34133-8]
- 126. Combination of the First-in-Class Imipridone ONC201 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468701/]
- 127. Mitochondrial unfolded protein response (UPRmt) as novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081383/]
- 128. The mitochondrial unfolded protein response (UPR mt ) [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-022-01317-0]
- 129. UPR, autophagy and mitochondria crosstalk underlies the ER ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4340752/]
- 130. Synergistic mechanism between the endoplasmic ... [https://www.nature.com/articles/s41420-023-01353-w]
- 131. Endoplasmic reticulum-mitochondria crosstalk: new ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1573499/full]
- 132. The Role of Endoplasmic Reticulum Stress and Unfolded ... [https://pubmed.ncbi.nlm.nih.gov/41089575/]
- 133. 急性髓系白血病新突破:UPR 相关基因特征助力精准诊疗 [https://www.ebiotrade.com/newsf/2025-2/20250226074419771.htm]
- 134. 基于未折叠蛋白反应(UPR)相关分子亚型的肝细胞癌免疫原性景观 ... [https://suppr.wilddata.cn/browses/detail/pubmed/37457742]
- 135. Targeting ER stress in skeletal muscle through physical activity [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582937/]
- 136. Endoplasmic Reticulum Stress in Neurodegenerative ... [https://www.mdpi.com/3042-4518/1/2/6]
- 137. The integrated stress response in neurodegenerative diseases [https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-025-00811-6]
- 138. Endoplasmic Reticulum-Associated Biomarkers for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7926397/]
- 139. An Impending Target for Multiple Neurological Disorders [https://www.frontiersin.org/research-topics/13977/unfolded-protein-response-upr-an-impending-target-for-multiple-neurological-disorders/magazine]
- 140. Modulation of Endoplasmic Reticulum Stress in ... [https://www.mdpi.com/1422-0067/26/13/6407]
- 141. The mitochondrial unfolded protein response: acting near ... [https://pubmed.ncbi.nlm.nih.gov/40237403/]
- 142. Recent advances in Alzheimer's disease: mechanisms ... [https://www.nature.com/articles/s41392-024-01911-3]
- 143. The Unfolded Protein Response—Novel Mechanisms ... [https://www.mdpi.com/2072-6694/17/22/3639]
- 144. In vivo manipulation of the protein homeostasis network ... [https://www.oncotarget.com/article/28764/text/]
- 145. Advances in Endoplasmic Reticulum Stress Research ... [https://www.mdpi.com/1422-0067/26/6/2487]