Protein Misfolding and Aggregation in Neurodegenerative Diseases: Mechanisms, Models, and Therapeutic Strategies
This article provides a comprehensive overview of the critical role of protein misfolding and aggregation in neurodegenerative diseases such as Alzheimer's, Parkinson's, and ALS.
Protein Misfolding and Aggregation in Neurodegenerative Diseases: Mechanisms, Models, and Therapeutic Strategies
Abstract
This article provides a comprehensive overview of the critical role of protein misfolding and aggregation in neurodegenerative diseases such as Alzheimer's, Parkinson's, and ALS. It explores the fundamental mechanisms of proteostasis collapse, oligomer toxicity, and prion-like propagation. For researchers and drug development professionals, the content details advanced methodological approaches including AI-driven prediction platforms and mathematical modeling. It further examines current challenges in therapeutic development, troubleshooting strategies for high-concentration formulations, and comparative analysis of therapeutic modalities targeting protein aggregation pathways. The synthesis of foundational science with cutting-edge applications offers a valuable resource for accelerating therapeutic innovation in this field.
The Proteostasis Crisis: Unraveling Core Mechanisms of Protein Misfolding in Neurodegeneration
Protein homeostasis, or proteostasis, is a fundamental biological process that maintains the cellular proteome in a functional state through an integrated system of synthesis, folding, trafficking, and degradation [1]. This exquisite balance ensures that proteins acquire and retain their correct three-dimensional structures, essential for executing myriad biological functions from catalytic activity to signal transduction [1] [2]. The proteostasis network (PN) represents approximately 3,000 genes that encode components working cooperatively across all three processes to provide surveillance of proteome integrity and limit toxic protein accumulation [3]. Disruption of this delicate balance—a state termed dysproteostasis—leads to protein misfolding, aggregation, and cellular dysfunction, underpinning a growing list of human diseases with particular significance in neurodegenerative disorders [4] [1].
The Proteostasis Network: Core Components and Functional Relationships
The PN encompasses three interconnected functional domains: protein synthesis, folding and trafficking, and degradation [3]. These components function collaboratively across nine organelle or process-specific branches: PN regulation and protein translation, nuclear, mitochondrial, endoplasmic reticulum (ER), extracellular and cytonuclear proteostasis, ubiquitin-proteasome system (UPS), and autophagy-lysosome pathway (ALP) [3].
Table 1: Core Components of the Proteostasis Network
| Functional Domain | Key Elements | Primary Functions |
|---|---|---|
| Protein Synthesis & Folding | Ribosomes, Heat Shock Proteins (HSPs: HSP70, HSP90, HSP60, small HSPs), Chaperonins (TRiC/CCT) | Nascent polypeptide folding; preventing aggregation; refolding misfolded proteins; protein trafficking [3] [1] [2] |
| Protein Degradation | Ubiquitin-Proteasome System (UPS), Autophagy-Lysosome Pathway (ALP), Chaperone-Mediated Autophagy (CMA) | Recognition and degradation of misfolded, damaged, or excess proteins [4] [3] [2] |
| Cellular Compartments & Stress Responses | Endoplasmic Reticulum (UPR), Mitochondrial UPR, Heat Shock Response (HSR) | Compartment-specific folding quality control; adaptive stress response activation [3] [1] [5] |
Diagram: The Integrated Proteostasis Network
Diagram Title: Integrated Proteostasis Network Components
Proteostasis Dysregulation in Neurodegenerative Diseases
In neurodegenerative diseases, collectively known as proteinopathies, proteostasis failure manifests through the accumulation of toxic, misfolded protein aggregates that lead to synaptic dysfunction and neuronal loss [4] [3]. The three most common dementias—Alzheimer's Disease (AD), Dementia with Lewy Bodies (DLB), and Frontotemporal Dementia (FTD)—are all characterized by distinct protein aggregates: AD by extracellular amyloid-beta (Aβ) plaques and intracellular hyperphosphorylated tau tangles; DLB by α-synuclein aggregates; and FTD by tau or TDP-43 proteins [4]. Aging represents the most significant risk factor for proteostasis decline, as chaperone expression and degradation efficiency progressively wane, rendering post-mitotic neurons particularly vulnerable to accumulated damage over a lifetime [4] [3].
Quantitative proteomic analyses of post-mortem human brains, animal models, and iPSC-derived neurons have confirmed that PN dysfunction is an early event in pathogenesis [4]. Mass spectrometry studies reveal that PN components heavily implicated in dementia pathogenesis include chaperones and the endolysosomal network (ELN), with ELN defects observed reproducibly and early in AD brains [4]. Genome-wide association studies further show enrichment of ELN proteins among AD risk factors [4].
Table 2: Proteostasis Network Associations in Human Diseases
| Disease Category | Proteostasis Proteins in Disease Gene Sets | Most Perturbed PN Pathways | Characteristic Proteostasis State |
|---|---|---|---|
| Neurodegenerative Diseases (AD, PD, FTD) | 30-35% | Autophagy-Lysosome Pathway (ALP), Ubiquitin-Proteasome System (UPS), Proteostasis Regulation | Extensive UPS + Extracellular Proteostasis perturbation [6] |
| Cancers (Lung, Kidney, Pancreatic) | 25-36% | UPS, Proteostasis Regulation | Significant UPS perturbation, limited extracellular proteostasis involvement [6] |
| Autoimmune, Cardiovascular, Endocrine | 15-25% | Extracellular Proteostasis, ALP | Distinctive extracellular proteostasis deregulation, limited UPS involvement [6] |
Quantitative Analysis of Proteostasis in Neurodegeneration
Advanced mass spectrometry (MS) technologies have enabled comprehensive quantitative analysis of the PN in dementia research. Johnson et al. utilized label-free quantification to study >2000 brains across different cohorts including AD, Asymptomatic AD (AsymAD), and non-AD controls [4]. Other studies have obtained deeper proteome coverage using extensive peptide fractionation techniques on smaller cohorts [4]. These approaches have been instrumental in distinguishing proteome changes that respond to protein aggregates from those responsible for cognitive deficits by comparing AsymAD patients (with Aβ and tau pathology but normal cognition) with Mild Cognitive Impairment (MCI) patients [4].
Key Experimental Findings
Chaperone Networks in AD: Inda et al. demonstrated that protein-protein interaction (PPI) chaperone networks were altered in AD human brains and mouse models using an affinity probe for stressed chaperones with label-free quantitation [4]. Spatial memory deficits were improved by inhibiting the formation of this stressed PPI network [4].
Chaperone-Mediated Autophagy (CMA): Bourdenx et al. used isobaric tags for quantitative proteomic analysis of insoluble brain fractions from CMA-deficient mice [4]. When crossed with AD mice, CMA deficiency enhanced insoluble proteins and exacerbated aggregation, whereas CMA activation decreased protein aggregation [4].
Endolysosomal Function: Lee et al. focused on SORLA, an endocytic receptor that mediates ELN trafficking [4]. Using isobaric tag quantitation on differentiated neurons from >50 iPSC lines of AD patients, they found SORLA expression significantly correlated with other AD risk factors localized to the ELN [4].
Diagram: Temporal Progression of Proteostasis Failure in Neurodegeneration vs. Cancer
Diagram Title: Temporal Proteostasis Perturbation Patterns
Experimental Methodologies for Proteostasis Research
Quantitative Proteomic Approaches
Mass Spectrometry (MS) Strategies:
- Label-Free Quantification: Used for large-scale studies (>2000 brains) to compare different cohorts (AD, AsymAD, Non-AD) [4]. This method measures peptide intensity without isotopic labeling, allowing analysis of numerous samples but with less accuracy than labeled methods.
- Isobaric Tag Quantitation (TMT/iTRAQ): Employed with extensive peptide fractionation for deeper proteome coverage in smaller cohorts [4]. Tags like tandem mass tags (TMT) enable multiplexing of samples, providing relative quantification across multiple conditions simultaneously.
- Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC): Metabolic labeling approach that incorporates heavy isotopes into proteins for accurate quantification, particularly useful in cell culture models.
Protein-Protein Interaction (PPI) Mapping
- Immunoprecipitation (IP) of Endogenous Proteins: Traditional method requiring specific antibodies to isolate protein complexes from brain tissue, followed by MS analysis [4].
- Affinity Probes for Stressed Chaperones: Specialized probes that selectively bind to chaperones under stress conditions, enabling isolation and quantification of stressed PPI networks [4].
- Proximity Labeling (BioID/APEX): Engineered enzymes that biotinylate nearby proteins, allowing identification of transient protein interactions in live cells [4]. Particularly valuable for mapping interactions of endogenous proteins in native environments like neurons.
Model Systems for Dementia Research
- Post-mortem Human Brain Tissue: Provides direct evidence but represents end-stage disease often with comorbidities [4].
- Transgenic Mouse Models: Overexpress human genes associated with dementia, enabling study of discrete timepoints but creating potential artifacts [4].
- Knock-in Models: Newer approaches without overexpression that better model the influence of aging on proteostasis decline [4].
- Induced Pluripotent Stem Cell (iPSC)-Derived Neurons: Patient-specific cells that can be differentiated into various neuronal and glial types, enabling drug screening combined with quantitative proteomics [4].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents for Proteostasis Network Studies
| Reagent/Material | Function/Application | Example Uses |
|---|---|---|
| Isobaric Tags (TMT, iTRAQ) | Multiplexed quantitative proteomics | Relative protein quantification across multiple samples in PN studies [4] |
| Stable Isotope Labels (SILAC) | Metabolic labeling for accurate quantification | Tracking protein synthesis and degradation dynamics [4] |
| Affinity Probes for Stressed Chaperones | Selective isolation of stressed PPI networks | Mapping altered chaperone interactions in disease models [4] |
| Proximity Labeling Enzymes (BioID, APEX) | Identification of transient protein interactions | Mapping endogenous PPI in neurons for PN components [4] |
| iPSC Lines | Patient-specific disease modeling | Differentiating into neuronal subtypes for cell-type-specific PN studies [4] |
| Specific Chaperone Inhibitors/Activators | Modulating specific PN pathways | Testing functional roles of HSP70, HSP90, CMA in protein aggregation [4] |
Therapeutic Targeting of Proteostasis Networks
Current therapeutic approaches focus on modulating PN components to ameliorate proteinopathies. Several strategies have shown promise in preclinical models:
- Chaperone Modulation: Small molecule inhibitors of stress-induced PPI networks have improved spatial memory in AD mouse models [4].
- CMA Activation: Enhancing chaperone-mediated autophagy decreased protein aggregation in AD models, while CMA deficiency exacerbated pathology [4].
- ELN-Targeted Approaches: Modulating endolysosomal trafficking through receptors like SORLA represents a potential avenue given the strong genetic association of ELN components with AD risk [4].
- HSF1 Activation: Enhancing the heat shock response to boost chaperone expression represents another strategy to combat proteostasis collapse [1] [2].
However, translating these approaches to human therapies has proven challenging. The modest efficacy of current AD drugs targeting Aβ aggregates, coupled with the abundance of AsymAD cases, suggests that protein aggregates are necessary but not sufficient to cause dementia [4]. This underscores the need for better understanding of PN dysfunction in early disease stages and developing therapeutics that target the underlying proteostasis imbalance rather than just its aggregated end products.
Future Perspectives and Research Directions
The future of PN research in neurodegenerative diseases requires several key developments:
- Direct PN Investigation: Quantitative proteomics needs to directly investigate the PN to understand its roles in the brain, aging, and dementia, using labeled strategies (heavy stable isotopes or isobaric tags) for better accuracy [4].
- Spatial Proteomics: Combining subcellular fractionation with PPI and post-translational modification analyses will determine how PN disruptions are distributed intracellularly [4].
- Cell-Type-Specific Resolution: New techniques to label cell-specific proteomes can quantify the cellular heterogeneity of the PN, understanding why certain neurons are more vulnerable [4].
- Comparative Studies: Quantitative proteomic comparisons of AD, FTD, and DLB are required to determine if therapeutic strategies can be universal or must be dementia-specific [4].
- Human-Relevant Models: Patient iPSCs differentiated into multiple neuronal and glial types will be essential for understanding cell-type-specific PN function and for drug screening [4].
The disappointing clinical trial outcomes for aggregate-targeting therapies in AD suggest that future success may require targeting earlier events in the proteostasis collapse cascade. As our understanding of PN architecture and regulation advances, particularly through quantitative proteomic approaches, new opportunities will emerge for therapeutic interventions that maintain or restore proteostasis balance in neurodegenerative diseases.
Key Misfolded Proteins in Major Neurodegenerative Disorders (Aβ, tau, α-synuclein, TDP-43, SOD1)
Neurodegenerative diseases represent one of the most significant challenges in modern medicine, with protein misfolding and aggregation serving as a central pathological hallmark across these conditions. More than 57 million people globally suffer from neurodegenerative diseases, a figure expected to double every 20 years [7]. The accumulation of misfolded proteins into toxic aggregates disrupts cellular homeostasis, triggers neuroinflammation, and ultimately leads to progressive neuronal loss [8] [9]. Despite their distinct clinical presentations, Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative conditions share common molecular mechanisms centered on protein misfolding [10] [11].
Understanding the structural properties, aggregation kinetics, and pathogenic mechanisms of key misfolded proteins—amyloid-β (Aβ), tau, α-synuclein, TDP-43, and SOD1—is crucial for developing targeted therapeutic strategies. These proteins, though functionally diverse in their native states, undergo conformational changes that expose hydrophobic regions, leading to the formation of soluble oligomers and ultimately insoluble fibrils and plaques [8] [9]. Recent research has highlighted the prion-like properties of these aggregates, enabling their propagation between cells and spreading pathology throughout connected brain regions [8] [10]. This whitepaper provides a comprehensive technical overview of these key misfolded proteins, their roles in disease pathogenesis, and the experimental approaches used to study them, framed within the context of protein misfolding and aggregation research.
Protein-Specific Pathogenesis and Characteristics
Table 1: Key Misfolded Proteins in Neurodegenerative Diseases
| Protein Name | Primary Associated Disease(s) | Aggregate Morphology & Key Characteristics | Genetic Loci | Key Pathogenic Forms |
|---|---|---|---|---|
| Amyloid-β (Aβ) | Alzheimer's disease (AD) | Extracellular senile plaques; Aβ42 more aggregation-prone than Aβ40 | APP, PSEN1, PSEN2 | Soluble oligomers, fibrils |
| Tau | AD, Pick's disease, PSP, CBD, AGD, CTE | Intracellular neurofibrillary tangles; hyperphosphorylated forms | MAPT | Oligomers, paired helical filaments |
| α-Synuclein | Parkinson's disease (PD), Dementia with Lewy Bodies (DLB), Multiple System Atrophy (MSA) | Neuronal Lewy bodies; glial cytoplasmic inclusions in MSA | SNCA | Soluble oligomers, protofibrils, fibrils |
| TDP-43 | ALS, FTLD, AD (30-70% of cases) | Cytoplasmic inclusions; nuclear clearance; hyperphosphorylation | TARDBP | Oligomers, aggregates, C-terminal fragments |
| SOD1 | Amyotrophic lateral sclerosis (ALS) | Cytoplasmic inclusions in motor neurons; dismutase activity loss | SOD1 | Misfolded monomers, aggregates |
| Huntingtin | Huntington's disease (HD) | Intracellular inclusions with expanded polyglutamine tract | HTT | N-terminal fragments, oligomers |
Table 2: Quantitative Properties of Misfolded Protein Aggregates
| Protein | Native Structure | Aggregation-Prone Domains | Critical Concentration for Aggregation | Half-Life in CSF/Plasma | Key Post-Translational Modifications |
|---|---|---|---|---|---|
| Aβ | Disordered monomer | Central hydrophobic cluster (CHC) | ~5-20 μM (Aβ42) | ~2 hours (plasma) | Phosphorylation, truncation |
| Tau | Disordered microtubule-binding protein | Microtubule-binding repeats | ~50-100 nM | ~3 weeks (CSF) | Hyperphosphorylation, acetylation, truncation |
| α-Synuclein | Disordered/N-terminal lipid-binding | NAC domain (residues 61-95) | ~50-100 μM | ~2 days (CSF) | Phosphorylation (S129), truncation, ubiquitination |
| TDP-43 | RNA-binding protein with RRM domains | Prion-like domain (C-terminal) | Not well characterized | ~16 hours (CSF) | Hyperphosphorylation, proteolytic cleavage |
| SOD1 | Stable homodimer | Electrostatic loop elements | Mutation-dependent | ~24 hours (CSF) | Oxidation, misfolding of metal-free form |
Amyloid-β (Aβ)
Aβ peptides are proteolytic fragments of the amyloid precursor protein (APP), produced through sequential cleavage by β- and γ-secretases [8] [12]. While Aβ40 is the most abundant isoform in the brain, the Aβ42 isoform, although produced in smaller quantities, is most prone to misfolding and aggregation, resulting in cytotoxic prefibrillar oligomers and fibrils that accumulate as extracellular senile plaques [9]. The structural transition of Aβ from random coil to β-sheet-rich conformations is driven by exposed hydrophobic regions, particularly in the central hydrophobic cluster (residues 17-21) and C-terminal domain [8]. Soluble Aβ oligomers are now regarded as primary drivers of neurotoxicity, disrupting synaptic function through multiple mechanisms including binding to neuronal receptors, inducing oxidative stress, and triggering inflammatory responses [8] [11].
Aβ pathology follows a distinct spatial-temporal progression, beginning in cortical regions and spreading throughout the brain in a prion-like manner [8]. The propagation of Aβ aggregates involves template-induced conformational change, where misfolded Aβ acts as a seed to convert native proteins into pathological forms. This process occurs at molecular, cellular, and organ scales, facilitating disease progression [8]. Recent evidence also indicates cross-seeding interactions between Aβ and other pathological proteins, particularly tau, exacerbating overall pathology [11] [13].
Tau Protein
Tau is a microtubule-associated protein that normally stabilizes microtubule networks in neuronal axons. In disease states, tau becomes hyperphosphorylated, misfolds, and aggregates into intracellular neurofibrillary tangles (NFTs) [8]. The transition from soluble to aggregated tau involves the formation of soluble oligomers that are highly toxic, followed by β-sheet-rich structures known as paired helical filaments (PHFs) that eventually form NFTs [8] [9]. Tau aggregation is promoted by numerous post-translational modifications, with hyperphosphorylation at specific epitopes (e.g., Ser202, Thr205, Thr231) serving as a key early event that reduces tau's affinity for microtubules and increases its aggregation propensity [13].
Tau pathology demonstrates a well-characterized progression through the brain, typically beginning in the transentorhinal region and advancing through hippocampal and cortical areas in a predictable pattern [8]. This spreading occurs through prion-like mechanisms where pathological tau seeds are released from affected cells and taken up by connected neurons, templating the misfolding of native tau in recipient cells [8] [10]. The molecular heterogeneity of tau aggregates contributes to different tauopathies, with distinct structural conformations or "strains" associated with specific diseases such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) [8].
α-Synuclein
α-Synuclein is a presynaptic protein that normally exists as an intrinsically disordered monomer but undergoes structural rearrangement to form β-sheet-rich aggregates that accumulate in Lewy bodies and Lewy neurites [8] [11]. The central non-amyloid-β component (NAC) region (residues 61-95) is critical for aggregation, with its hydrophobic nature driving the initial oligomerization [8]. The pathogenesis of α-synuclein in Parkinson's disease unfolds as a multistep process, initiating with protein misfolding that leads to the formation of increasingly intricate oligomers, soluble intermediates, and eventually insoluble fibrils and aggregates [11].
The prion-like propagation of α-synuclein pathology follows a characteristic pattern through the nervous system, beginning in the dorsal motor nucleus of the vagus and olfactory bulb, then spreading through the brainstem to midbrain and cortical regions [8]. This spreading occurs through cell-to-cell transmission involving the release of α-synuclein aggregates from donor cells, their uptake by recipient cells, and subsequent seeding of new aggregates [8] [10]. The aggregates of α-synuclein have the potential to induce atrophy through diverse mechanisms, encompassing lysosomal impairment, mitochondrial dysfunction, endoplasmic reticulum stress, and dysfunction in synaptic transmission [11].
TDP-43
TDP-43 is an RNA/DNA binding protein that normally resides predominantly in the nucleus but in disease conditions mislocalizes to the cytoplasm where it forms hyperphosphorylated and ubiquitinated aggregates [8] [13]. The protein contains two RNA recognition motifs (RRM1 and RRM2) and a C-terminal domain with a prion-like glycine-rich region that mediates protein-protein interactions and drives aggregation [13]. Pathological TDP-43 is characterized by hyperphosphorylation (particularly at Ser409/410), proteolytic cleavage generating C-terminal fragments (~25-35 kDa), and loss of nuclear function [13].
In approximately 30-70% of Alzheimer's disease cases, TDP-43 co-aggregates with Aβ and tau, with patients exhibiting more severe cognitive impairment, faster disease progression, and greater hippocampal atrophy [13]. TDP-43 interacts with both major AD pathologies: it promotes Aβ oligomerization and directly influences tau pathology by regulating tau mRNA stability and alternative splicing [13]. TDP-43 also disrupts mitochondrial function, RNA metabolism, and protein quality control, creating a vicious cycle of proteostatic collapse [9] [13].
SOD1
Superoxide dismutase 1 (SOD1) is a homodimeric metalloenzyme that catalyzes the conversion of superoxide radicals to oxygen and hydrogen peroxide. Mutations in the SOD1 gene, particularly those that destabilize the native structure or reduce metal binding, promote misfolding and aggregation in amyotrophic lateral sclerosis [10] [12]. Misfolded SOD1 species lose their dismutase activity and acquire toxic gain-of-function properties, including pro-oxidant activity, mitochondrial dysfunction, and impairment of protein quality control systems [10].
SOD1 aggregates propagate in a prion-like manner, with misfolded SOD1 acting as a template to convert native SOD1 into pathological forms [8] [10]. These aggregates can spread between cells and induce misfolding of native SOD1, highlighting the role of prion-like mechanisms in the progression of ALS [8]. The accumulation of SOD1 aggregates disrupts proteostasis by saturating chaperone systems and impairing proteasomal and autophagic clearance pathways, leading to widespread protein dyshomeostasis [10].
Common Aggregation Mechanisms and Cross-Talk
Despite their structural differences, the pathogenic proteins share common aggregation mechanisms including nucleation-dependent polymerization, where the formation of an initial seed represents the rate-limiting step, followed by rapid elongation and amplification [8] [9]. Each protein can form multiple structurally distinct aggregates or "strains" that exhibit different pathological properties, seeding activities, and disease phenotypes [8]. This conformational diversity may explain the heterogeneity in clinical presentations and disease progression rates observed in patients.
Growing evidence indicates significant cross-talk between different pathological proteins. The accumulation of one misfolded protein can impair the entire proteostatic network, triggering the misfolding of unrelated proteins that would otherwise fold normally [10]. For instance, Aβ can promote tau hyperphosphorylation and aggregation, while TDP-43 pathology interacts with both Aβ and tau to exacerbate Alzheimer's pathology [11] [13]. These interactions create vicious cycles that accelerate disease progression and complicate therapeutic targeting.
Diagram 1: Protein Misfolding and Propagation. The diagram illustrates the molecular events in protein misfolding, from initial native structure to oligomer formation, fibril elongation, and intercellular propagation through seeding. Toxic oligomers (yellow) directly cause neuronal dysfunction, while the cyclic process of fragmentation and templating enables disease progression.
Research Methodologies and Experimental Protocols
Aggregate Detection and Characterization
Immunoassay-Based Detection (ELISA/Simoa) Protocol Principle: Sandwich immunoassays enable specific detection and quantification of target proteins and their aggregated forms in biological fluids and tissue extracts. Detailed Methodology:
- Coating: Coat microplate wells with capture antibodies specific for target protein epitopes (e.g., 6E10 for Aβ, AT8 for phosphorylated tau, Syn211 for α-synuclein).
- Blocking: Block nonspecific binding sites with protein-based blockers (5% BSA or non-fat dry milk in PBS-Tween).
- Sample Preparation: Dilute CSF/plasma/tissue homogenates in appropriate buffers. For aggregate-specific detection, use native conditions without denaturation.
- Incubation: Add samples and standards to wells, incubate 2-4 hours at room temperature with shaking.
- Detection: Add biotinylated detection antibody (targeting distinct epitope), incubate 1-2 hours.
- Signal Development: Add streptavidin-HRP conjugate, incubate 30-60 minutes, then add chemiluminescent or colorimetric substrate.
- Quantification: Measure signal intensity and calculate concentrations from standard curves. Technical Notes: For ultrasensitive detection, use Single Molecule Array (Simoa) technology with paramagnetic beads and enzyme-labeled detection antibodies for digital counting of individual molecules [14].
Seeding Amplification Assays (RT-QuIC) Protocol Principle: Real-Time Quaking-Induced Conversion assays exploit the prion-like seeding activity of protein aggregates to amplify minute quantities for detection. Detailed Methodology:
- Recombinant Substrate: Express and purify recombinant full-length target protein (e.g., α-synuclein, tau) in E. coli.
- Sample Preparation: Dilute CSF/brain homogenate in PBS with 0.1% SDS to dissociate non-specific aggregates while preserving seeding-competent species.
- Reaction Setup: Mix sample with recombinant substrate (0.1-0.3 mg/mL) in black 96-well plates with glass beads.
- Cyclic Incubation: Incubate plates at 37-42°C with intermittent shaking cycles (e.g., 1 minute shaking, 14 minutes rest).
- Thioflavin T Monitoring: Include 10-20 μM Thioflavin T, monitor fluorescence (excitation 450 nm, emission 480 nm) after each cycle.
- Data Analysis: Determine amplification time and maximum fluorescence; compare to standards for semi-quantification. Technical Notes: Assay conditions must be optimized for each protein target; α-synuclein RT-QuIC typically requires 150-200 cycles, while tau assays may need different buffer conditions [8] [14].
Cellular and Animal Models
Primary Neuronal Cultures for Toxicity Assessment Protocol Principle: Utilize primary neurons to evaluate the direct neurotoxic effects of protein aggregates and screen therapeutic compounds. Detailed Methodology:
- Cortical/Hippocampal Dissection: Isolate brain regions from E16-18 rodent embryos or postnatal day 0-1 pups.
- Tissue Dissociation: Digest tissue with papain or trypsin, triturate to single-cell suspension.
- Culture Establishment: Plate cells on poly-D-lysine-coated plates in neurobasal medium with B27 supplement, glutamine, and penicillin/streptomycin.
- Treatment Preparation: Prepare recombinant oligomers (Aβ, α-synuclein, tau) by agitating monomer solutions at 300-1000 rpm for 12-48 hours; characterize by AFM/TEM and SEC.
- Compound Testing: Pre-treat neurons with experimental compounds 2-24 hours before adding protein aggregates.
- Viability Assessment: After 24-72 hours exposure, measure cell viability using MTT reduction, LDH release, or live/dead staining.
- Functional Assays: Assess mitochondrial membrane potential (JC-1 dye), reactive oxygen species (DCFDA), synaptic density (immunostaining for PSD-95, synapsin). Technical Notes: Use 7-14 days in vitro (DIV) neurons for maturity; include both monomer and vehicle controls [8] [11].
Transgenic Mouse Models Protocol Principle: Engineer mice to express human mutant proteins to recapitulate key aspects of protein aggregation pathology. Detailed Methodology:
- Model Selection: Choose appropriate models - APP/PS1 for Aβ pathology, P301S for tauopathy, A53T for α-synuclein, SOD1G93A for ALS.
- Genotyping: Confirm transgene presence by PCR of tail DNA.
- Intervention Studies: Administer test compounds via oral gavage, intraperitoneal injection, or intracerebroventricular infusion starting pre-symptomatically or at early disease stages.
- Behavioral Assessment: Conduct motor tests (rotarod, open field) and cognitive tests (Morris water maze, Y-maze) at regular intervals.
- Tissue Collection: Perfuse animals transcardially with PBS, collect brains and spinal cords for fixed (4% PFA) and fresh-frozen processing.
- Pathological Analysis: Perform immunohistochemistry for protein aggregates, stereological cell counting, biochemical fractionation for soluble/insoluble proteins. Technical Notes: Include age-matched non-transgenic and vehicle-treated controls; power studies appropriately for group sizes (n=10-15) [10] [11].
Table 3: Essential Research Reagents and Resources
| Reagent Category | Specific Examples | Key Applications | Technical Considerations |
|---|---|---|---|
| Antibodies | 6E10 (Aβ), AT8/Tau5 (tau), Syn211 (α-syn), 2E2-D3 (TDP-43 pS409/410) | IHC, WB, ELISA, immunoprecipitation | Validate specificity for aggregated vs. monomeric forms |
| Assay Kits | Human Aβ42 ELISA, p-tau181 Simoa, α-syn RT-QuIC, TDP-43 MSD | Biomarker quantification, aggregate detection | Consider matrix effects in biological fluids |
| Cell Lines | SH-SY5Y, HEK293, BV-2 microglia, primary neuronal cultures | Toxicity screening, mechanism studies | Use relevant models (primary neurons for physiological relevance) |
| Animal Models | APP/PS1 (AD), P301S (tauopathy), A53T (α-syn), SOD1G93A (ALS) | Pathogenesis studies, therapeutic testing | Account for species differences in protein sequences |
| Recombinant Proteins | Monomeric Aβ1-42, α-synuclein, tau (2N4R, 1N4R), TDP-43 full-length | Biophysical studies, seeding assays | Ensure proper purification and characterization of conformation |
Biomarker Applications and Therapeutic Strategies
Table 4: Biomarker and Therapeutic Development Pipeline
| Target/Pathway | Biomarker Status | Therapeutic Approaches | Clinical Trial Status |
|---|---|---|---|
| Aβ aggregation | Plasma Aβ42/40 ratio (CLIA-approved) | Monoclonal antibodies (Lecanemab, Donanemab), BACE inhibitors | Phase 3 completed/ongoing |
| Tau pathology | CSF p-tau181, p-tau217 (validated) | Anti-tau antibodies, tau aggregation inhibitors, ASOs | Phase 2-3 trials |
| α-Synuclein aggregation | RT-QuIC (CSF, tissue), seed amplification assays | Immunotherapies, ASOs, small molecule inhibitors | Phase 1-2 studies |
| TDP-43 pathology | Plasma NfL (non-specific), CSF TDP-43 fragments | RNA-targeting therapies, modulation of nucleocytoplasmic transport | Preclinical/early clinical |
| Microglial function | sTREM2 (CSF), microglial PET imaging | TREM2 agonists (AL002, VG-3927), CD33 modulation | Phase 1-2 trials |
Biomarker Development
Fluid biomarkers have transformed neurodegenerative disease research and clinical trials by providing objective measures of pathological processes [14] [7]. Blood-based biomarkers offer particular promise for scalable screening and longitudinal monitoring. The most established biomarkers include:
- Aβ42/Aβ40 ratio: Decreased ratio indicates amyloid pathology; measured using immunoprecipitation-mass spectrometry or immunoassays [14]
- Phosphorylated tau (p-tau181, p-tau217): Highly specific for Alzheimer's tau pathology; strong correlation with tau PET [14]
- Neurofilament light chain (NfL): Non-specific marker of neuroaxonal damage; elevated across multiple neurodegenerative conditions [14] [7]
- sTREM2: Marker of microglial activation; shows dynamic changes during disease progression [15]
Recent technological advances including single-molecule array (Simoa) and mass spectrometry-based methods have enabled precise quantification of these biomarkers in blood, overcoming previous sensitivity limitations [14]. The Global Neurodegeneration Proteomics Consortium (GNPC) has established one of the world's largest harmonized proteomic datasets, including approximately 250 million unique protein measurements from over 35,000 biofluid samples, accelerating biomarker discovery and validation [7].
Therapeutic Strategies Targeting Protein Aggregation
Immunotherapy Approaches Monoclonal antibodies represent the most advanced therapeutic strategy targeting protein aggregates. Lecanemab, Aducanumab, and Donanemab target Aβ aggregates with varying epitope specificities, demonstrating clearance of amyloid plaques [15]. These antibodies work primarily by enhancing microglial phagocytosis of aggregates through Fc receptor engagement [15]. Similar approaches are being developed for α-synuclein (Prasinezumab) and tau (Gosuranemab, Tilavonemab) [11]. Challenges include limited blood-brain barrier penetration, potential inflammatory side effects (ARIA with anti-Aβ antibodies), and targeting of appropriate protein species [15].
Small Molecule Inhibitors Small molecules targeting various stages of the aggregation cascade include:
- Aggregation inhibitors: Compounds like epigallocatechin gallate (EGCG) and CLR01 that block the initial oligomerization steps
- Protein clearance enhancers: Activators of autophagy (rapamycin analogs) or the ubiquitin-proteasome system
- Chaperone inducers: Compounds that boost cellular chaperone networks (arimoclomol, celastrol)
- Kinase inhibitors: Modulators of kinases involved in pathological phosphorylation (GSK-3β, CK1δ inhibitors for tau) [11] [13]
Novel Mechanistic Approaches Emerging strategies include:
- Antisense oligonucleotides (ASOs): Reduce production of target proteins; Tofersen for SOD1-ALS shows clinical benefit [10]
- Gene therapy: AAV-mediated delivery of protective factors (BDNF, PGRN) or knockdown of mutant genes
- Modulation of protein quality control systems: Enhancement of proteasome function, autophagy, or the unfolded protein response [11]
- Microglial-targeted therapies: TREM2 agonists (AL002, VG-3927), CD33 antagonists that enhance clearance of protein aggregates [15]
Diagram 2: Therapeutic Targeting of Protein Aggregation. The diagram illustrates key intervention points in the protein aggregation cascade, including ASOs to reduce synthesis, small molecules to inhibit oligomerization, immunotherapies to clear fibrils, and microglial modulators to address neuroinflammation. Enhancing proteostasis machinery (green) represents another key therapeutic strategy.
The study of misfolded proteins in neurodegenerative diseases has progressed dramatically from initial histopathological observations to sophisticated molecular understanding of aggregation mechanisms and their consequences. Key challenges remain, including the need for biomarkers that can detect pathology in the earliest stages, therapies that can effectively target the most toxic species, and strategies to address the heterogeneity of these diseases.
Future research directions should focus on:
- Early intervention strategies: Targeting pathological processes before irreversible neuronal loss occurs
- Combination therapies: Addressing multiple aspects of the disease cascade simultaneously
- Patient stratification: Using biomarker profiles to match patients with optimal treatments
- Novel therapeutic modalities: Developing approaches that target RNA, enhance protein clearance, or modulate neuroinflammation
- Advanced delivery systems: Ensuring therapeutics reach the appropriate brain regions and cell types
The integration of multi-omics technologies, advanced imaging, and sophisticated cellular models will continue to deepen our understanding of these complex diseases. International collaborative efforts like the Global Neurodegeneration Proteomics Consortium are essential for assembling the large datasets needed to identify robust biomarkers and therapeutic targets [7]. As our knowledge of protein misfolding mechanisms grows, so does the potential for developing effective treatments that can alter the course of these devastating disorders.
The seeding-nucleation model of protein aggregation represents a fundamental mechanism underlying the pathogenesis of numerous neurodegenerative diseases. This process involves the transition of native monomeric proteins into toxic oligomeric species and eventually into amyloid fibrils, driven by a nucleation-dependent polymerization mechanism. The formation of stable nucleation seeds constitutes the rate-limiting step, after which rapid elongation and amplification occur through prion-like template-directed misfolding. This comprehensive review examines the molecular mechanisms, kinetic principles, and experimental methodologies for studying protein aggregation, with particular emphasis on the central role of oligomeric intermediates in neurotoxicity. Emerging therapeutic strategies targeting specific stages of the aggregation cascade are also discussed, providing a framework for developing interventions against protein misfolding disorders.
Protein aggregation following the seeding-nucleation model is a central pathological feature in neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) [16] [17]. These conditions are characterized by the accumulation of specific misfolded proteins—such as amyloid-β (Aβ) and tau in AD, α-synuclein (α-syn) in PD, and huntingtin (Htt) in HD—that form protein aggregates disrupting cellular homeostasis and ultimately leading to neuronal death [16] [17] [18].
The seeding-nucleation mechanism was first described for prion proteins (PrP) and has since been recognized as a fundamental process underlying the aggregation kinetics of numerous amyloidogenic proteins implicated in neurodegeneration [19] [18]. This model explains the characteristic lag phase observed during in vitro aggregation assays, where soluble monomers slowly form stable nucleation seeds, followed by a rapid growth phase where these seeds template the conversion of additional monomers into fibrillar structures [19] [18].
Understanding the molecular events governing the transition from monomers to oligomers is particularly crucial, as accumulating evidence indicates that soluble oligomeric species—rather than mature fibrils—represent the primary mediators of neurotoxicity in protein misfolding disorders [17] [18]. These oligomers exhibit heightened toxicity through multiple mechanisms, including membrane disruption, mitochondrial dysfunction, impairment of proteostatic systems, and induction of oxidative stress [18].
This review comprehensively examines the seeding-nucleation model of protein aggregation within the context of neurodegenerative disease research, with emphasis on molecular mechanisms, experimental approaches, and therapeutic implications.
Molecular Mechanisms of Seeding-Nucleation
The Kinetic Pathway of Aggregation
The aggregation of amyloidogenic proteins follows a characteristic kinetic trajectory comprising three distinct phases: lag phase, elongation phase, and plateau phase. During the initial lag phase, soluble monomers undergo conformational changes and associate into unstable oligomeric intermediates until critical nuclei form. This nucleation step is rate-limiting and stochastic, explaining the variable onset of aggregation observed experimentally [18]. Once stable seeds are established, the process enters a rapid elongation phase where these seeds act as templates for the sequential addition of monomers, leading to exponential growth of fibrillar structures. Finally, the system reaches a plateau phase as the available monomer pool depletes and equilibrium establishes between soluble and insoluble species [17] [18].
The diagram below illustrates the sequential stages of the seeding-nucleation model:
Oligomeric Intermediates: Structure and Toxicity
Oligomeric assemblies formed during the aggregation process represent crucial intermediates with distinct structural and toxic properties. These species are typically characterized by their amorphous structures with exposed hydrophobic regions, making them highly reactive and cytotoxic [17]. Oligomers exist in a dynamic equilibrium with monomers and fibrils, and can be categorized as either "on-pathway" (progressing toward fibril formation) or "off-pathway" (terminal products that do not convert to fibrils) [17].
The structural transition from disordered monomers to β-sheet-rich oligomers constitutes a critical step in the aggregation process. In environments with limited solvent availability, such as those mimicked by reverse micelles, monomeric amyloid-β (Aβ) proteins can form extended β-strands, providing a plausible mechanism for amyloid fibril nucleation in the brain [17]. This structural transformation is initiated by the formation of β-strand seed structures that act as nucleation sites for further aggregation.
Multiple studies have demonstrated that soluble oligomers from various amyloidogenic proteins (Aβ, α-syn, tau) exhibit greater correlation with disease progression and neuronal dysfunction than mature fibrils or insoluble deposits [17] [18]. The mechanisms underlying oligomer toxicity include:
- Membrane disruption through pore formation or bilayer distortion
- Mitochondrial dysfunction and increased reactive oxygen species (ROS) production
- Impairment of synaptic plasticity and neuronal signaling
- Disruption of proteostasis by overwhelming protein quality control systems
- Induction of inflammatory responses in glial cells
Table 1: Characteristics of Oligomeric Species in Neurodegenerative Diseases
| Protein | Size Range | Key Structural Features | Primary Toxic Mechanisms |
|---|---|---|---|
| Aβ | 10-50 monomers | Prefibrillar assemblies with exposed hydrophobic patches | Membrane disruption, synaptic dysfunction, oxidative stress |
| α-Synuclein | 10-30 monomers | Annular protofibrils with β-sheet content | Mitochondrial impairment, membrane permeability, vesicle trafficking disruption |
| Tau | 10-40 monomers | Aggregates with cross-β structure | Microtubule destabilization, impaired axonal transport |
| Huntingtin | 10-100 monomers | Spherical and annular structures with polyQ expansion | Proteasome inhibition, transcription dysregulation, organelle dysfunction |
Prion-like Propagation and Cell-to-Cell Transmission
A defining characteristic of many pathogenic protein aggregates is their capacity for prion-like propagation, enabling the spread of pathology throughout the brain [19] [17] [18]. This process involves the release of misfolded proteins from donor cells, their uptake by recipient cells, and subsequent seeding of new aggregates in previously healthy cells [19] [17].
The molecular mechanisms facilitating cell-to-cell transmission include:
- Non-classical secretion of aggregated proteins via exocytotic vesicles [19]
- Exosome-mediated transfer of pathogenic species between cells [19] [17]
- Receptor-mediated endocytosis of extracellular aggregates [19]
- Direct penetration of plasma membranes by oligomeric species [19]
The following diagram illustrates the prion-like propagation mechanism:
The seeding efficiency of pathogenic aggregates depends on multiple factors, including their structural properties (strain characteristics), cellular environment, and the presence of co-factors that can either promote or inhibit the process [18]. Different conformational variants (strains) of the same protein can exhibit distinct seeding activities and neuropathological profiles, potentially explaining the heterogeneity in clinical presentations and disease progression observed among patients with the same neuropathological diagnosis [17] [18].
Experimental Methods and Research Tools
Label-Free Detection of Protein Aggregates
Traditional approaches for visualizing protein aggregates rely on fluorescent tags (e.g., GFP, YFP, mCherry) fused to the protein of interest. However, these tags can significantly alter the biophysical properties of proteins, including aggregation kinetics, final aggregate structure, and interactions with cellular components [20]. For instance, GFP-tagged huntingtin exon 1 (Httex1) forms fibrils approximately 3 nm thicker than untagged Httex1 and exhibits altered mechanical properties and interactomes [20].
To address these limitations, label-free methods have been developed that enable the study of unaltered protein aggregates:
- Label-free Identification of NDD-associated Aggregates (LINA): Utilizes deep learning to detect unlabeled protein aggregates in living cells from transmitted-light images, allowing quantitative analysis of aggregation dynamics without fluorescent labeling [20]
- Quantitative Phase Imaging (QPI): Produces quantitative images of unlabeled specimens based on phase shifts, enabling extraction of parameters such as dry mass and morphology with minimal photodamage [20]
- Brillouin Microscopy: A non-invasive technique that studies biomechanical properties of cells and tissues through inelastic scattering, revealing that protein aggregates exhibit higher longitudinal modulus compared to cytoplasm [21]
- Self-driving microscopy: Employs deep learning to predict aggregation onset from images of soluble protein, achieving 91% accuracy in triggering optimized multimodal imaging when aggregation is imminent [21]
The experimental workflow for label-free aggregate detection is summarized below:
Quantitative Assessment of Aggregation Kinetics
The table below summarizes key parameters and methods for quantifying protein aggregation kinetics:
Table 2: Quantitative Parameters for Protein Aggregation Analysis
| Parameter | Description | Measurement Techniques | Typical Values/Ranges |
|---|---|---|---|
| Lag Phase Duration | Time required for nucleation seed formation | Thioflavin T fluorescence, light scattering | Minutes to hours (in vitro); days in cellular models |
| Elongation Rate | Speed of fibril growth after nucleation | Atomic force microscopy, TEM with time-lapse | Variable depending on protein and conditions |
| Oligomer Concentration | Amount of soluble oligomeric species | ELISA, native PAGE, photo-induced cross-linking | Nanomolar to low micromolar range in disease models |
| Seeding Efficiency | Potency in inducing aggregation in recipient cells | FRET-based biosensors, cell-based seeding assays | Strain-dependent; affected by aggregate size and structure |
| Toxicity Threshold | Oligomer concentration causing cell death | Viability assays, membrane integrity tests | Protein-specific; often correlates with oligomer size |
Research Reagent Solutions
The table below outlines essential research tools and reagents for studying protein aggregation:
Table 3: Essential Research Reagents for Protein Aggregation Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Recombinant Proteins | Aβ1-42, α-synuclein, tau isoforms | In vitro aggregation studies, seeding assays | Source, purification method, endotoxin levels affect reproducibility |
| Pre-formed Fibrils (PFFs) | α-syn PFFs, tau PFFs | Seeding experiments in cellular and animal models | Preparation method, sonication protocol, concentration |
| Fluorescent Tags | GFP, mCherry, SNAP-tag | Visualizing aggregation in live cells | Tags can alter native aggregation kinetics and properties |
| Aggregation Sensors | Thioflavin T, Congo red, Proteostat | Detecting amyloid formation in vitro and in cells | Specificity, background signal, compatibility with live cells |
| Cell Lines | SH-SY5Y, HEK293, primary neurons | Cellular models of protein aggregation | Species origin, differentiation state, transfection efficiency |
| Animal Models | APP/PS1 mice, Thy1-α-syn mice, HD knock-in | In vivo study of aggregation and spread | Genetic background, age of onset, pathological progression |
Experimental Protocols
Protocol: Seeding Assay Using Pre-formed Fibrils (PFFs)
Purpose: To evaluate the seeding activity of pre-formed fibrils on endogenous soluble proteins in cellular models.
Materials:
- Purified pre-formed fibrils (PFFs) of protein of interest (e.g., α-syn, tau)
- Target cell line (e.g., HEK293 cells, primary neurons)
- Lipofectamine or alternative transfection reagent (for some protocols)
- Fixation and immunostaining reagents
- Confocal microscopy equipment
Procedure:
- Prepare PFFs by sonicating fibril stocks (3 × 10 seconds pulses at 10-20% amplitude) to generate short fragments suitable for cellular uptake.
- Treat cells with PFFs at appropriate concentration (typically 0.1-5 μg/mL) for 4-24 hours.
- Remove extracellular PFFs by washing with PBS and maintain cells in fresh media for desired time period (typically 3-14 days).
- Fix cells and perform immunostaining for protein of interest and aggregation markers (e.g., phospho-epitopes).
- Image using confocal microscopy and quantify percentage of cells with aggregates, aggregate number per cell, and aggregate size.
Key Considerations: PFF concentration, sonication parameters, and cell type significantly impact seeding efficiency. Include appropriate controls (monomer-treated cells, vehicle-only) in each experiment [19].
Protocol: Label-Free Identification of Aggregates in Living Cells
Purpose: To detect and quantify protein aggregates in living cells without fluorescent labeling.
Materials:
- Cells expressing protein of interest (without fluorescent tag)
- Microscope with transmitted-light or phase-contrast capability
- LINA software and pre-trained neural network models [20]
- Computer with appropriate GPU for deep learning analysis
Procedure:
- Culture cells expressing untagged protein of interest under appropriate conditions.
- Acquire transmitted-light image stacks at multiple z-planes using microscope.
- Process images using Fourier filtering to generate quantitative phase images (QPI).
- Apply LINA deep learning model to identify aggregates from phase images.
- Quantify aggregate parameters including number, area, dry mass, and growth kinetics.
Key Considerations: The method is robust across imaging conditions and different protein constructs, providing high-speed, specific identification of native aggregates without tag-induced alterations [20].
Therapeutic Implications and Future Directions
The seeding-nucleation model provides critical insights for developing therapeutic strategies targeting protein aggregation in neurodegenerative diseases. Current approaches include:
- Seeding inhibitors: Compounds that prevent the template-directed conversion of native proteins (e.g., small molecules that bind to oligomeric species) [16] [17]
- Passive immunotherapy: Antibodies targeting toxic oligomeric species to neutralize their toxicity and promote clearance [16] [17]
- Enhancement of protein clearance: Activation of autophagy-lysosomal pathway or ubiquitin-proteasome system to degrade aggregates [16]
- Gene therapy: Reduction of mutant protein expression using antisense oligonucleotides or RNA interference [16] [17]
Understanding the precise molecular mechanisms underlying the formation and propagation of pathogenic seeds continues to drive innovation in diagnostic and therapeutic development for neurodegenerative proteinopathies. The emergence of advanced imaging technologies and label-free detection methods promises to accelerate these efforts by providing more physiologically relevant insights into protein aggregation dynamics.
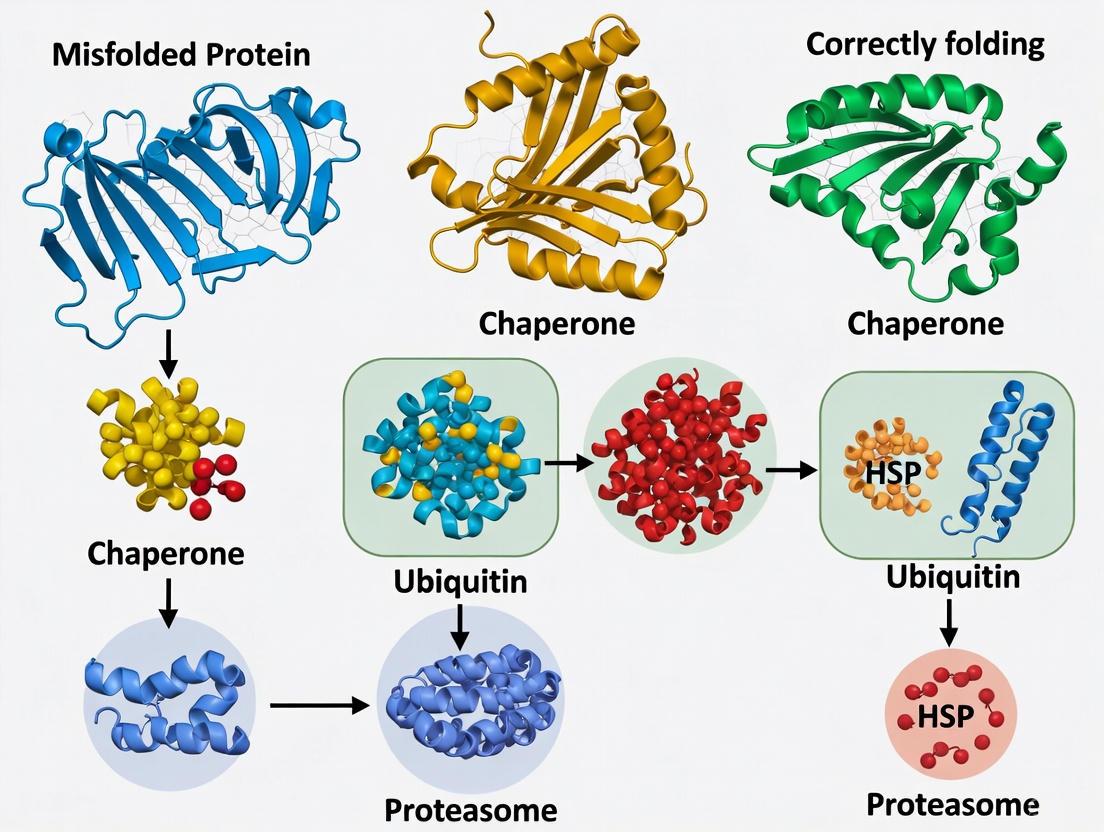
Protein homeostasis, or proteostasis, is essential for neuronal function and survival. It encompasses the coordinated cellular processes that regulate protein synthesis, folding, trafficking, and degradation [10] [22]. The ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP) represent the two major intracellular protein degradation systems responsible for maintaining this delicate balance [23] [24]. In neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Amyotrophic Lateral Sclerosis (ALS), the accumulation of misfolded and aggregated proteins is a defining pathological feature [10] [16] [11]. This failure in proteostasis is often attributed to impairments in the UPS and ALP, which become overwhelmed by the burden of aberrant proteins or are directly compromised by the disease process itself [23] [10] [11]. A comprehensive understanding of these clearance pathways, their intricate crosstalk, and their role in neurodegeneration is paramount for developing novel therapeutic strategies aimed at restoring proteostatic control and neuronal health.
The Ubiquitin-Proteasome System (UPS)
Molecular Mechanism and Components
The UPS is a highly specific, ATP-dependent proteolytic system responsible for the degradation of the majority of intracellular soluble proteins, particularly short-lived and soluble misfolded proteins [23] [25]. It operates through a coordinated two-step process: the covalent attachment of ubiquitin chains to a target protein, followed by its recognition and degradation by the proteasome [22] [24].
Ubiquitination is a enzymatic cascade involving three key components:
- E1 (Ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent manner [24] [25].
- E2 (Ubiquitin-conjugating enzyme): Accepts the activated ubiquitin from E1 [24] [25].
- E3 (Ubiquitin ligase): Recognizes specific substrate proteins and facilitates the transfer of ubiquitin from E2 to the target protein, conferring substrate specificity. The human genome encodes hundreds of E3 ligases, allowing for precise targeting [22] [24] [25].
Proteins tagged with chains of ubiquitin linked through lysine 48 (K48) are typically directed to the proteasome for degradation [25]. The proteasome is a multi-subunit complex comprising a 20S core particle (CP) and one or two 19S regulatory particles (RP) [22]. The 19S RP recognizes ubiquitinated proteins, removes the ubiquitin chains, and unfolds the substrate. The unfolded polypeptide is then translocated into the 20S CP, a barrel-shaped structure containing proteolytic active sites that hydrolyze the protein into short peptides [22] [24].
Figure 1: The Ubiquitin-Proteasome System Pathway. This diagram illustrates the sequential enzymatic cascade of ubiquitination and subsequent proteasomal degradation of a target protein.
The UPS in Neurodegenerative Diseases
Impairment of the UPS is heavily implicated in the pathogenesis of various neurodegenerative diseases. Protein aggregates enriched with ubiquitinated proteins are a hallmark of these disorders, suggesting a failure in UPS-mediated clearance [10] [11]. In cerebral ischemia, for example, ubiquitinated proteins accumulate, and proteasome function is compromised [23]. The depletion of free ubiquitin and the accumulation of its mutant form can directly inhibit the UPS, contributing to delayed neuronal death [23].
Specific disease-associated proteins are also linked to UPS dysfunction:
- In Parkinson's disease, mutations in E3 ubiquitin ligases like Parkin are associated with familial forms of the disease. Parkin promotes the degradation of proteins like Drp1, and its loss of function exacerbates neuronal injury [23].
- In Alzheimer's disease, components of the UPS are found associated with neurofibrillary tangles and amyloid plaques [10].
- The immunoproteasome, a specialized proteasome induced during inflammation, is upregulated in cerebral ischemia. Its inhibition has been shown to reduce infarction volume and attenuate inflammation, suggesting a role in ischemic pathology [23].
Table 1: Key UPS Components and Their Alterations in Neurodegenerative Pathologies
| UPS Component | Normal Function | Alteration in Disease | Associated Diseases |
|---|---|---|---|
| E3 Ligase (Parkin) | Substrate ubiquitination | Loss-of-function mutations | Parkinson's Disease [23] |
| E3 Ligase (TRAF6) | Signal transduction | Overexpression exacerbates injury | Cerebral Ischemia [23] |
| Immunoproteasome | Immune response, stress clearance | Subunit (β1i, β5i) upregulation | Cerebral Ischemia, Stroke [23] |
| Deubiquitinase (USP14) | Negatively regulates proteasome | Inhibition is neuroprotective | Cerebral Ischemia [23] |
| Ubiquitin C-terminal Hydrolase L1 (UCHL1) | Deubiquitination | Mutation (C152A) is protective | Ischemic Stroke [23] |
The Autophagy-Lysosome Pathway (ALP)
Molecular Mechanism and Forms of Autophagy
The ALP is responsible for the degradation of long-lived proteins, insoluble protein aggregates, and damaged organelles [23] [25]. It is a vesicular trafficking pathway that delivers cargo to the lysosome for degradation. There are three main forms of autophagy:
- Macroautophagy: The primary form, involving the formation of a double-membraned vesicle, the autophagosome, which engulfs cytoplasmic cargo. The autophagosome then fuses with the lysosome to form an autolysosome, where the contents are degraded by acidic hydrolases [24] [25].
- Microautophagy: The direct engulfment of cytoplasmic material by invagination of the lysosomal membrane.
- Chaperone-Mediated Autophagy (CMA): A highly selective process where specific proteins bearing a KFERQ-like motif are recognized by chaperones (Hsc70) and directly translocated across the lysosomal membrane via the LAMP-2A receptor for degradation [11].
Lysosomes can also receive extracellular material and cell-surface receptors via endocytosis, phagocytosis, and pinocytosis [24] [25].
Figure 2: The Macroautophagy Pathway. This diagram illustrates the key stages of macroautophagy, from the formation of the phagophore to the degradation of cargo within the autolysosome.
The ALP in Neurodegenerative Diseases
Similar to the UPS, the ALP is critically impaired in neurodegenerative diseases. The accumulation of autophagic vesicles is a common observation in affected neurons, indicating a blockage in the autophagic flux [11] [26]. Many disease-linked aggregate-prone proteins, such as mutant huntingtin (in HD) and α-synuclein (in PD), are substrates for autophagy, and their clearance is heavily dependent on a functional ALP [10] [11].
Specific disease connections include:
- In Parkinson's disease, α-synuclein aggregates can impair CMA by binding to and obstructing the LAMP-2A receptor, thereby inhibiting its own degradation and that of other CMA substrates [11].
- In Alzheimer's disease, defective autophagy contributes to the accumulation of Aβ peptides and hyperphosphorylated tau. Enhancing autophagy has been shown to reduce pathology in animal models [11] [26].
- In cerebral ischemia, the autophagy-lysosome pathway is impaired, contributing to neuronal damage [23].
Table 2: Autophagy-Lysosome Pathway Impairments in Neurodegenerative Diseases
| Disease | Key Aggregated Protein(s) | ALP Impairment |
|---|---|---|
| Alzheimer's Disease (AD) | Amyloid-β, Tau | Reduced autophagosome clearance, disrupted lysosomal function [11] [26] |
| Parkinson's Disease (PD) | α-Synuclein | Inhibition of Chaperone-Mediated Autophagy (CMA), impaired mitophagy [11] |
| Huntington's Disease (HD) | Mutant Huntingtin (mHtt) | Defective cargo recognition and autophagosome transport [10] |
| Amyotrophic Lateral Sclerosis (ALS) | TDP-43, SOD1 | Impaired autophagic flux, lysosomal dysfunction [10] [12] |
| Cerebral Ischemia | Various ubiquitinated proteins | Impaired autophagy-lysosome pathway [23] |
Crosstalk and Therapeutic Targeting
Interplay Between UPS and ALP
The UPS and ALP are not isolated systems but are interconnected and collaborate to maintain proteostasis. When one pathway is compromised, the other can often compensate. For instance, proteasome inhibition can induce the upregulation of autophagy [26]. Conversely, impairment in autophagy can lead to increased dependence on the UPS. This crosstalk becomes critical under conditions of cellular stress, such as in neurodegenerative diseases where the burden of misfolded proteins overwhelms both systems, leading to a vicious cycle of proteostatic collapse [23] [11] [26]. The Keap1-Nrf2-ARE signaling pathway is one example of a regulatory node that can be activated by proteotoxic stress and can transcriptionally upregulate components of both degradation systems [11].
Experimental Protocols for Studying Clearance Pathways
Protocol 1: Assessing Proteasome Activity in Cell Cultures
- Cell Treatment: Treat cultured neuronal cells (e.g., SH-SY5Y, primary neurons) with a proteasome inhibitor (e.g., MG-132, 10 μM) as a negative control or a potential activator (e.g, Trehalose) for a defined period (e.g., 6-24 h) [23] [10].
- Cell Lysis: Lyse cells in a buffer containing ATP to maintain 26S proteasome integrity.
- Activity Assay: Use fluorogenic peptides that are substrates for different proteasome catalytic activities (e.g., Suc-LLVY-AMC for chymotrypsin-like activity). Incubate cell lysates with the substrate and measure the release of the fluorescent group (AMC) over time using a microplate reader [22].
- Data Analysis: Normalize fluorescence values to total protein concentration. Activity is expressed as fluorescence intensity/hour/mg of protein.
Protocol 2: Monitoring Autophagic Flux Using LC3-I/II Turnover
- Transfection/Treatment: Transfect cells with a GFP-LC3 plasmid or treat cells with an autophagy modulator (e.g., Rapamycin for induction, Bafilomycin A1 for inhibition of lysosomal degradation) [24].
- Protein Extraction and Western Blot: Harvest cells and extract proteins. Perform Western blot analysis using antibodies against LC3.
- Detection and Quantification: LC3-I (cytosolic form) and the phosphatidylethanolamine-conjugated LC3-II (autophagosome-associated form) will be detected. An increase in LC3-II levels in the presence of a lysosomal inhibitor (like Bafilomycin A1) indicates increased autophagic flux, whereas an increase without inhibition suggests a blockade in the pathway [24] [25].
- Immunofluorescence: Alternatively, visualize GFP-LC3 puncta (representing autophagosomes) under a confocal microscope. An increase in puncta number indicates autophagy induction or blockade of downstream steps.
Protocol 3: Evaluating Protein Aggregation via Filter Trap Assay
- Sample Preparation: Prepare solubilized protein extracts from brain tissue or cultured cells using buffers with mild detergents.
- Filtration: Dilute samples and filter through a cellulose acetate membrane under vacuum. Soluble proteins pass through, while large, insoluble aggregates are trapped.
- Immunodetection: Probe the membrane with an antibody specific to the protein of interest (e.g., anti-α-synuclein for PD, anti-polyglutamine for HD).
- Analysis: Detect the signal using chemiluminescence. The amount of trapped aggregate correlates with the signal intensity [10].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Studying Protein Clearance Pathways
| Reagent / Assay | Function / Application | Example Use in Research |
|---|---|---|
| Fluorogenic Proteasome Substrates (e.g., Suc-LLVY-AMC) | Quantifying chymotrypsin-like proteasome activity in cell lysates or purified complexes. | Measuring proteasome dysfunction in cerebral ischemia models [23] [22]. |
| LC3B Antibody | Detecting LC3-I to LC3-II conversion via Western blot or immunofluorescence to monitor autophagosome formation. | Assessing autophagic flux in neurons under oxidative stress [24]. |
| p62/SQSTM1 Antibody | Detecting p62, an autophagy receptor that is degraded along with its cargo; accumulation indicates autophagy impairment. | Demonstrating blocked autophagic degradation in disease models [24] [11]. |
| Lysosomal Trackers & pHrodo Dyes | Staining acidic organelles (lysosomes, autolysosomes) and tracking endocytosis based on vesicle acidification. | Studying lysosomal function and vesicle trafficking in live cells [24]. |
| PROTAC Molecules (Heterobifunctional degraders) | Inducing targeted degradation of specific proteins of interest (POIs) via the UPS for therapeutic exploration. | Degrading pathological tau or mutant huntingtin in preclinical studies [24] [25]. |
| Bafilomycin A1 | V-ATPase inhibitor that blocks autophagosome-lysosome fusion and lysosomal acidification. | Used to inhibit autophagic degradation and measure autophagic flux [24]. |
Emerging Therapeutic Strategies
Therapeutic strategies aimed at enhancing protein clearance are a major focus of neurodegenerative disease research. These include:
UPS-Targeted Therapies: Small molecule proteasome activators are being explored to boost the degradation of misfolded proteins. Additionally, targeted protein degradation (TPD) technologies, such as PROteolysis TArgeting Chimeras (PROTACs), are being developed to artificially recruit specific disease-causing proteins to E3 ubiquitin ligases for ubiquitination and degradation [25]. For example, PROTACs have been designed to target the androgen receptor and estrogen receptor in cancer, and this approach is now being applied to neurodegenerative disease targets [25].
ALP-Targeted Therapies: Pharmacological inducers of autophagy, such as Trehalose and Rapamycin analogs (e.g., Everolimus), have shown promise in preclinical models by enhancing the clearance of aggregate-prone proteins and improving neuronal survival [23] [11]. Trehalose has been shown to inhibit cerebral ischemia-induced protein aggregation by preserving proteasome activity [23] [10].
Lysosome-Targeting Therapies: For extracellular proteins and cell-surface receptors, LYsosome-TArgeting Chimeras (LYTACs) represent a novel modality that binds to both the extracellular protein and a lysosome-targeting receptor, shuttling the target for lysosomal degradation [24] [25].
Multi-Target Approaches: Given the crosstalk between pathways, interventions that simultaneously enhance both UPS and ALP function, such as activating the transcription factor TFEB (a master regulator of lysosomal biogenesis and autophagy), are under intense investigation [11].
Molecular chaperones, particularly heat shock proteins (HSPs), constitute a primary cellular defense mechanism against proteotoxic stress by ensuring proper protein folding, preventing aberrant aggregation, and facilitating the degradation of irreversibly damaged proteins. Within the context of neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), the collapse of this chaperone system contributes directly to the accumulation of toxic protein aggregates, a hallmark of these disorders. This whitepaper provides an in-depth analysis of chaperone classification, structure, and mechanistic functions in protein homeostasis. It further examines the consequences of chaperone network failure in neurodegeneration and details contemporary experimental methodologies for investigating chaperone-client interactions. Finally, we synthesize current therapeutic strategies aimed at augmenting chaperone function to combat protein misfolding diseases, presenting quantitative data on drug development pipelines and reagent solutions for research professionals.
Cellular protein homeostasis, or proteostasis, represents a delicate equilibrium between protein synthesis, folding, trafficking, and degradation [27]. This balance is maintained by a sophisticated network of molecular chaperones, folding enzymes, and degradation machineries. Molecular chaperones, a class of highly conserved proteins, are fundamental components of this proteostasis network (PN) [28]. They function as cellular defense proteins by interacting with, stabilizing, and assisting other proteins in acquiring their functionally active native conformations, thereby preventing misfolding and aggregation [28] [29]. The term "heat shock protein" originates from their initial discovery in Drosophila as proteins markedly upregulated in response to thermal stress [27] [28]. However, their expression and function are critical under physiological conditions for managing nascent polypeptide chains and under various proteotoxic stresses, including oxidative stress and metabolic alterations [30] [29].
The challenge of maintaining proteostasis is particularly acute in neurons. As post-mitotic cells, neurons cannot dilute accumulated toxic aggregates through cell division, making them exceptionally vulnerable to proteostasis imbalances over time [29]. A hallmark of numerous neurodegenerative diseases is the intra- or extracellular accumulation of misfolded proteins that aggregate into toxic oligomers and insoluble fibrils, leading to synaptic dysfunction, activation of stress pathways, and ultimately neuronal death [10] [31] [11]. Key aggregating proteins include amyloid-β (Aβ) and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, and huntingtin with expanded polyglutamine tracts in Huntington's disease [10] [12]. Molecular chaperones are deployed at every stage to combat this pathogenic cascade, from initial folding assistance to refolding, sequestration, and ultimate degradation of misfolded species [31] [29]. The failure of these defense mechanisms is a pivotal event in the progression of neurodegeneration, positioning chaperones as central players in both disease pathogenesis and potential therapeutic strategies [10] [28] [11].
Classification, Structure, and Mechanism of Molecular Chaperones
Molecular chaperones are systematically classified based on their molecular weight, structure, and mechanism of action. The major families include HSP100, HSP90, HSP70, HSP60, HSP40, and small HSPs (sHSPs), alongside regulatory co-chaperones that fine-tune their activity [28] [29].
Table 1: Major Molecular Chaperone Families and Their Characteristics
| Chaperone Family | Key Members | ATP-Dependent | Cellular Localization | Primary Functions |
|---|---|---|---|---|
| Small HSPs (sHSPs) | HSPB1 (HSP27), HSPB5 | No | Cytosol, Nucleus | First line of defense; prevent aggregation by binding unfolding clients [28] |
| HSP40 (DnaJ) | DnaJA, DnaJB, DnaJC | Co-chaperone | Cytosol, Nucleus | Regulate HSP70 ATPase activity; client protein recruitment [28] |
| HSP70 | Hsp70, Hsc70 | Yes | Cytosol, Nucleus, ER, Mitochondria | Nascent chain folding; prevent aggregation; refold misfolded proteins; client delivery to degradative pathways [32] [29] |
| HSP90 | Hsp90α, Hsp90β, GRP94, TRAP-1 | Yes | Cytosol, ER, Mitochondria | Maturation of specific client proteins (e.g., kinases, transcription factors) [28] |
| HSP60/HSP10 | Chaperonins | Yes | Mitochondria, Cytosol | Facilitate folding in sequestered chambers, especially for complex proteins [28] |
Structural Insights and the Chaperone Cycle
Structural biology has profoundly advanced our understanding of chaperone mechanisms. The first crystal structures of HSP family members, including HSC70 (1993) and the N-terminal domain of HSP90 (1997), paved the way for elucidating their functional cycles [28]. Recent breakthroughs include resolving structures of ternary and quaternary complexes, such as the HSP90-CDC37-kinase and HSP90-HSP70-HOP-GR complexes, revealing the intricate mechanics of client protein regulation [28].
The HSP70 cycle serves as a paradigm for ATP-dependent chaperone function. HSP70 comprises an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD). In the ATP-bound state, the SBD is open, allowing for rapid binding and release of client proteins with exposed hydrophobic regions. HSP40 co-chaperones, which themselves can bind clients, stimulate ATP hydrolysis by HSP70. This hydrolysis traps the client in the SBD. Subsequent nucleotide exchange, often facilitated by co-chaperones like HSP110, promotes ADP release and ATP rebinding, leading to client release. This cycle allows HSP70 to bind unfolded polypeptides, prevent aggregation, and facilitate folding [28] [32] [29].
The following diagram illustrates the core ATP-dependent cycle of HSP70 chaperones:
Similary, the HSP90 chaperone machinery involves a complex and dynamic process. HSP90 functions as a flexible dimer, and its cycle is regulated by a suite of co-chaperones like CDC37, p23, and Aha1. The cycle progresses through distinct conformational states (open, closed) that are coupled to ATP binding and hydrolysis. Co-chaperones act as molecular switches that dictate client recruitment, activation, or degradation. For instance, the co-chaperone CDC37 specifically recruits kinase clients to HSP90, forming a transient ternary complex that is essential for kinase stability and maturation [28].
Chaperone Functions in Neurodegeneration: Mechanisms and Consequences
In neurodegenerative proteinopathies, molecular chaperones engage in a multi-layered defense strategy against proteotoxic stress, targeting the misfolded proteins associated with these diseases.
Anti-Aggregation and Refolding
The primary function of chaperones is to bind to hydrophobic patches exposed on misfolded proteins, shielding them from inappropriate interactions that lead to aggregation. sHSPs like HSP27 act as a first line of defense by forming large oligomers that bind unfolding clients, preventing aggregation in an ATP-independent manner [28]. ATP-dependent chaperones like HSP70 and HSP40 then attempt to refold these stabilized clients. If initial refolding fails, the Hsp70-Hsp40 system, sometimes with the assistance of Hsp110 in metazoans, can function as a disaggregase, actively disentangling and solubilizing pre-formed aggregates to generate refoldable species [29].
Clearance of Misfolded Proteins
When refolding attempts are unsuccessful, chaperones pivot to facilitate the degradation of terminally misfolded proteins by partnering with cellular clearance machinery.
- Ubiquitin-Proteasome System (UPS): Hsp70 can recognize misfolded clients and present them to E3 ubiquitin ligases, which tag the clients for processive degradation by the 26S proteasome [29].
- Autophagy-Lysosome Pathways: Chaperones are integral to multiple forms of autophagy.
- Chaperone-Mediated Autophagy (CMA): Hsc70 (a constitutive HSP70 isoform) recognizes proteins bearing a KFERQ-like motif and directs them to the lysosomal membrane receptor LAMP2A for translocation into the lysosome and degradation [31] [29].
- Macroautophagy: Misfolded proteins and aggregates that are too large for the proteasome or CMA are tagged by chaperones and selectively engulfed by autophagosomes via adaptor proteins like p62/SQSTM1. The autophagosome then fuses with the lysosome for bulk degradation [11] [29].
The following pathway diagram synthesizes the major chaperone-mediated defense mechanisms against proteotoxic stress:
Chaperone Dysfunction in Disease
Aging and chronic proteotoxic stress can lead to a state of "proteostatic collapse," where chaperone networks become overwhelmed and dysfunctional [30]. In many neurodegenerative diseases, components of the chaperone system are found sequestered within insoluble protein aggregates, rendering them inactive and further exacerbating the proteostasis imbalance [31] [32]. For example, in Parkinson's disease, α-synuclein can inhibit CMA by binding to LAMP2A, thereby blocking its own degradation and creating a vicious cycle of accumulation [31]. Similarly, in Alzheimer's and Huntington's disease, a specific subset of chaperones linked to protein synthesis (CLIPs) is repressed, weakening the cell's ability to manage newly synthesized proteins and existing misfolded species [10].
Experimental Methodologies for Chaperone Research
Investigating the structure, function, and client interactions of molecular chaperones requires a multidisciplinary approach. The following table outlines key reagent solutions and methodologies essential for this field.
Table 2: Research Reagent Solutions for Molecular Chaperone Studies
| Research Tool | Specific Examples | Function/Application | Key Experimental Use |
|---|---|---|---|
| Recombinant Chaperones | Human Hsp90β, Hsp70, Hsp40 | Purified proteins for in vitro assays | Study ATPase activity, client binding, and refolding kinetics without cellular complexity [28] |
| Small Molecule Inhibitors/Activators | HSP90: Geldanamycin derivatives; HSP70: MAL3-101 | Chemically modulate chaperone function | Validate chaperones as drug targets; probe mechanism by disrupting specific functions in cells and animal models [28] [33] |
| Antibodies for Detection | Anti-Hsp70, Anti-Hsp90, Anti-tau, Anti-α-synuclein | Detect expression, localization, and post-translational modifications | Immunoblotting, immunofluorescence, immunohistochemistry to assess levels and co-localization in models and patient samples [12] |
| Cell-Based Assay Systems | FRET-based reporters; Luciferase refolding assays | Monitor protein folding, aggregation, and chaperone activity in live cells | High-throughput screening for chaperone modulators; study real-time proteostasis dynamics [12] |
| Microplate Reader Assays | ELISA, Thioflavin T (ThT) aggregation assays | Quantify protein aggregation and misfolding | Measure kinetics of amyloid formation and the inhibitory effects of chaperones or drugs [12] |
A generalized workflow for a key experiment investigating chaperone inhibition and its effect on client protein stability is outlined below. This is commonly used to validate chaperone-client relationships and the efficacy of novel inhibitors.
Detailed Protocol: Assessing Client Protein Degradation upon HSP90 Inhibition
- Cell Treatment: Culture an appropriate cell line (e.g., a cancer cell line known to express HSP90-dependent kinase clients). Treat with a titrated dose of a well-characterized HSP90 inhibitor (e.g., 17-AAG) or a vehicle control (DMSO). A protein synthesis inhibitor like cycloheximide can be added to isolate the effect on protein degradation.
- Lysate Collection: Harvest cells at predetermined time points (e.g., 0, 2, 4, 8, 16 hours) post-inhibition. Lyse cells using RIPA buffer supplemented with protease and phosphatase inhibitors to preserve post-translational modifications.
- Immunoblot Analysis: Resolve equal amounts of protein by SDS-PAGE and transfer to a membrane. Probe with antibodies against known HSP90 client proteins (e.g., HER2, AKT, CDK4) and the chaperone itself. A decrease in client protein levels over time, without a corresponding decrease in a non-client loading control (e.g., Actin), indicates client degradation upon chaperone inhibition.
- Phenotypic Validation: Analyze parallel samples for functional consequences, such as reduced cell viability (MTT assay), induction of apoptosis (caspase-3/7 activity), or cell cycle arrest (flow cytometry) [28].
Therapeutic Targeting and Clinical Outlook
The critical role of chaperones in neurodegenerative diseases makes them attractive therapeutic targets. Strategies range from boosting the entire chaperone network to developing highly specific small-molecule modulators.
Table 3: Stages of Therapeutic Development Targeting Molecular Chaperones
| Development Stage | Targeting Strategy | Example Agents/Candidates | Key Opportunities | Key Challenges |
|---|---|---|---|---|
| Stage 1: Pan-Isoform Inhibition | Broad inhibition of all isoforms within an HSP family | HSP90: Geldanamycin, 17-AAG | Validated HSP90 as a drug target, particularly in oncology [28] [33] | Toxicity due to lack of selectivity; compensatory induction of other HSPs [33] |
| Stage 2: Isoform-Selective Inhibition | Targeting specific isoforms (e.g., cytosolic vs. ER) | HSP90: Selective inhibitors for GRP94 or TRAP-1 | Potential for reduced toxicity and tissue-specific targeting [28] | Structural similarity between isoforms makes high selectivity difficult to achieve [33] |
| Stage 3: Protein-Protein Interaction (PPI) Inhibition | Disrupting specific chaperone-co-chaperone-client interactions | Inhibitors of Hsp90-CDC37 for kinase-driven cancers | Higher specificity, potentially bypassing compensatory HSP induction [28] | PPIs often involve large, shallow surfaces, making drug design challenging [28] [33] |
| Stage 4: Multi-Specific Molecules & Novel Modalities | Bifunctional degraders (PROTACs); HSP expression inducers | Hsp70-based PROTACs; compounds that activate HSF1 | Potential to achieve synergistic effects; targeted protein degradation [28] [33] | Complex chemistry and pharmacology; long-term safety unknown |
While most clinical progress has been in oncology, strategies for neurodegeneration are advancing. These include:
- HSP Inducers: Compounds that activate the heat shock transcription factor 1 (HSF1) to upregulate a broad spectrum of HSPs, thereby reinforcing the proteostasis network [10].
- CMA Enhancers: Small molecules that stabilize the LAMP2A receptor or otherwise enhance CMA flux to promote clearance of α-synuclein and tau [31] [11].
- Natural Products: Molecules like curcumin have been shown to modulate HSP expression and possess anti-aggregation properties, offering multi-target potential [31].
A significant challenge in neurodegeneration is the identification of biomarkers to monitor disease progression and therapeutic response, which is crucial for the success of clinical trials [10]. Collaborative ecosystems involving academia, industry, and patient advocacy groups are essential to accelerate the development of these novel chaperone-targeting therapies.
Molecular chaperones stand as a fundamental cellular bulwark against proteotoxic stress, their function being paramount to neuronal health and viability. The detailed mechanistic understanding of their structure, functional cycles, and roles in protein quality control has illuminated their central contribution to the pathogenesis of neurodegenerative proteinopathies. The continued elucidation of complex chaperone-client interactions, coupled with advances in structural biology and rational drug design, is opening new avenues for therapeutic intervention. Targeting molecular chaperones—whether by enhancing their refolding capacity, boosting clearance pathways, or inhibiting specific pathological interactions—holds immense promise for developing disease-modifying treatments for Alzheimer's, Parkinson's, Huntington's, and other related disorders. For researchers and drug development professionals, the future lies in innovating beyond traditional inhibition towards sophisticated, multi-specific strategies that can restore proteostatic balance to the aging or diseased brain.
The propagation of misfolded proteins across cellular networks in the brain is a seminal feature of many neurodegenerative diseases. This process, termed "prion-like propagation," describes the cell-to-cell transmission of protein aggregates and the templated seeding of their normally folded endogenous counterparts in recipient cells [34] [18]. Unlike prion diseases, which are infectious, these common neurodegenerative diseases (e.g., Alzheimer's disease (AD), Parkinson's disease (PD), and frontotemporal dementia) show no evidence of interindividual transmission, yet the spatial and temporal progression of pathology in the brain is highly stereotyped [34] [35]. This whitepaper delineates the molecular mechanisms underlying this propagation, details key experimental methodologies for its study, and discusses implications for therapeutic development within the broader context of protein misfolding and aggregation research.
Core Mechanisms of Prion-like Propagation
The prion-like propagation of protein pathology is not a random process but a multi-step cascade that enables the disease to spread through neuroanatomically connected circuits.
The Conformational Template and Seeding
The fundamental event in prion-like propagation is a templated conformational change. A misfolded protein, acting as a "seed" or "nucleating particle," induces the native, soluble protein to adopt its own abnormal conformation [34] [36]. This seed can be an oligomer, protofibril, or mature fibril. The newly misfolded protein can then integrate into the growing aggregate, and the process repeats, leading to the exponential amplification of pathology within a cell [18] [36].
Table 1: Key Protein Propagons in Neurodegenerative Diseases
| Disease | Misfolded Protein | Primary Inclusion Type | Seeding Competence |
|---|---|---|---|
| Alzheimer's Disease (AD) | Amyloid-β (Aβ) & Tau | Plaques & Neurofibrillary Tangles (NFTs) | Demonstrated for both Aβ and Tau in vivo [35] [18] |
| Parkinson's Disease (PD) | α-Synuclein | Lewy Bodies | Yes; human brain-derived extracts can seed pathology in mice [37] |
| Frontotemporal Dementia (FTD) | Tau (3R or 4R isoforms) | Neuronal & Glial Inclusions | Yes; distinct conformers (strains) possible [34] [38] |
| Multiple System Atrophy (MSA) | α-Synuclein | Glial Cytoplasmic Inclusions | Yes; highly potent seeding activity shown [37] |
| Amyotrophic Lateral Sclerosis (ALS) | TDP-43 | Cytoplasmic Inclusions | Prion-like seeding and spreading observed [18] |
Cellular Uptake and Release
For propagation to occur, seeds must exit the "donor cell" and enter a "recipient cell."
- Release Mechanisms: Pathological seeds can be released into the extracellular space through several mechanisms, including non-conventional exocytosis, active secretion via exosomes, or through membrane rupture associated with cell death [36].
- Uptake Mechanisms: Once extracellular, seeds can be internalized by neighbouring neurons or glial cells through endocytosis, macropinocytosis, or via specific receptors on the cell surface [38].
Network Spread
The spread of pathology does not occur randomly but follows synaptic and anatomical connections [34]. For example, in AD, tau pathology originates in the transentorhinal region, progresses to the hippocampus and limbic areas, and finally reaches the neocortex [35]. This pattern of spread has been corroborated by functional imaging studies and is consistent with the prion-like propagation of tau aggregates along neural networks [34].
Figure 1: The Cyclic Process of Prion-like Propagation. The diagram illustrates the multi-step mechanism by which a misfolded protein seed leads to the spread of pathology through a neuronal network.
Strain Diversity and Selective Vulnerability
A critical concept from prion biology that applies to other neurodegenerative diseases is that of "strains." Distinct, self-propagating conformations of the same protein can give rise to different disease phenotypes, including variations in clinical presentation, disease progression, and neuropathological profile [34]. For instance, different tau filament structures, determined by cryo-electron microscopy, underpin the unique neuropathological signatures of AD, Pick's disease, and chronic traumatic encephalopathy [35] [38].
This strain phenomenon may also underlie selective neuronal vulnerability. Recent research indicates that certain types of neurons may be more susceptible to the propagation of specific protein strains. A 2024 study demonstrated that the 22L prion strain propagates more efficiently in glutamatergic excitatory neurons than in GABAergic inhibitory neurons in the mouse brain [39]. This neuronal cell-type-dependent tropism provides a mechanistic basis for the regional distribution of pathology observed in many neurodegenerative conditions.
Table 2: Factors Influencing Strain Properties and Neuronal Vulnerability
| Factor | Impact on Propagation | Experimental Evidence |
|---|---|---|
| Protein Conformation | Dictates seeding efficiency, stability, and toxicity. | Distinct Aβ fibril conformers show different toxicities in primary neurons [34]. |
| Post-Translational Modifications (e.g., Phosphorylation) | Can modulate aggregation propensity and seeding activity. | Hyperphosphorylation of tau is a hallmark of tauopathies and affects its aggregation [35] [38]. |
| Neuronal Cell Type | Differences in gene expression and proteostasis network can affect susceptibility. | 22L prion strain shows preferential propagation in Vglut1/Vglut2+ excitatory neurons [39]. |
| Innate Immune Response (Microglia/Astrocytes) | Can either clear aggregates or contribute to neuroinflammation and spread. | Microglial activation is deeply involved in prion disease neuropathology [39]. |
Key Experimental Models and Protocols
Validating the prion-like hypothesis and developing therapeutics requires robust experimental models that recapitulate the seeding and propagation of protein pathology.
In Vivo Seeding and Propagation Models
The gold-standard experiment involves the intracerebral injection of pathological seeds into a model animal to monitor the de novo induction and spread of protein aggregates.
Protocol: Intrastriatal Injection of Human Brain-Derived α-Synuclein Seeds in Mice [37]
- Seed Preparation: Insoluble protein fractions are extracted from frozen cortical tissue of human MSA or control patients.
- Homogenize tissue in high-salt buffer (e.g., 10 mM Tris-HCl, 0.8 M NaCl) with protease/phosphatase inhibitors.
- Add sarkosyl (2% final concentration) to solubilize membranes and isolate insoluble aggregates.
- Centrifuge at high speed (e.g., 100,000 × g) to pellet the sarkosyl-insoluble fraction, which is enriched for pathological seeds.
- Resuspend the pellet in PBS for injection.
- Stereotaxic Surgery:
- Anesthetize recipient mice (e.g., mice expressing human wild-type α-synuclein).
- Fix the animal in a stereotaxic frame and perform a craniotomy.
- Inject the seed preparation (e.g., 30 μL) into the left striatum using precise coordinates relative to the Bregma.
- Analysis:
- Sacrifice animals at multiple time points (e.g., 3, 6, 9 months) post-injection.
- Analyze brains using immunohistochemistry for phosphorylated α-synuclein to detect induced pathology.
- Perform biochemical analyses (e.g., ELISA, Western blot) to confirm the presence of hyperphosphorylated, insoluble α-synuclein.
Outcome: Mice injected with MSA brain extracts, but not controls, develop phosphorylated α-synuclein inclusions that begin in the injected hemisphere and progressively spread to connected, contralateral regions over time [37].
Figure 2: In Vivo Seeding Assay Workflow. The experimental pipeline for assessing the seeding and propagation capability of human brain-derived pathological proteins in a mouse model.
In Vitro Seeding Assays
Cell-based and biochemical assays allow for high-throughput screening of seeding inhibitors and detailed mechanistic studies.
- FRET-Based Biosensor Cell Lines: These cells express the disease protein of interest (e.g., tau) fused to cyan and yellow fluorescent proteins. Upon seeded aggregation, the fluorophores come close enough for FRET to occur, providing a quantifiable signal [18].
- Thioflavin T (ThT) Binding Assay: This biochemical assay uses the dye Thioflavin T, which exhibits enhanced fluorescence upon binding to the cross-β-sheet structure of amyloid fibrils. It is used to monitor the kinetics of fibril growth in real-time when seeds are added to soluble protein monomers [37].
The Scientist's Toolkit: Key Research Reagents
The following table compiles essential reagents and models critical for conducting research in prion-like propagation.
Table 3: Essential Research Reagents and Resources
| Reagent / Model | Function and Utility | Key Characteristics |
|---|---|---|
| Sarkosyl-Insufficient Fractions | Enrichment of pathological, aggregate-prone proteins from human or animal brain tissue. | Key step in preparing bioactive seeds for in vivo and in vitro experiments [37]. |
| Tg(SNCA)1Nbm/J Mice | A mouse model expressing human wild-type α-synuclein on a mouse α-synuclein knockout background. | Crucial for demonstrating human protein-dependent propagation without interference from mouse protein [37]. |
| FRET-Based Biosensor Cell Lines | Live-cell reporting of protein aggregation seeded by exogenous material. | Enables high-throughput screening of therapeutic compounds that inhibit seeding [18]. |
| Phospho-Specific Antibodies (e.g., AT8 for tau) | Immunodetection of disease-associated, hyperphosphorylated protein forms in tissue and blots. | Allows for the specific visualization and quantification of pathology spread [37] [38]. |
| Recombinant Misfolded Protein Fibrils | Defined, synthetic seeds for controlled experimentation without requiring human tissue. | Useful for mechanistic studies, though may not fully replicate the heterogeneity of patient-derived seeds [18]. |
Implications for Therapeutic Development
Understanding prion-like propagation opens novel therapeutic avenues aimed at halting disease progression by targeting the spread of pathology.
- Immunotherapy: Both active and passive immunization strategies are being explored to generate antibodies that target extracellular seeds, facilitating their clearance by the immune system before they can be internalized by recipient cells [35] [36].
- Seeding Inhibitors: Small molecules or peptides that stabilize the native protein conformation or directly block the interaction between the seed and the native protein could prevent the templated misfolding cascade [36].
- Enhanced Protein Clearance: Boosting cellular proteostasis networks, including the autophagy-lysosomal pathway and the ubiquitin-proteasome system, could help clear seeds and aggregates once they have formed inside the cell [36].
A significant challenge is the strain diversity of pathological aggregates, which may necessitate therapies that are tailored to specific conformations or that target a common structural motif shared by all oligomeric species [18].
The prion-like propagation mechanism provides a powerful paradigm for explaining the progressive nature of major neurodegenerative diseases. The intercellular transfer of pathological seeds and the subsequent templated corruption of native proteins drive the relentless spread of pathology through the brain. This framework not only advances our fundamental understanding of disease pathogenesis but also directly informs the development of a new class of disease-modifying therapies. Future research must focus on characterizing the precise nature of the toxic seeds in human brain, understanding the determinants of selective neuronal vulnerability, and translating anti-propagation strategies into effective clinical treatments.
Advanced Methodologies: AI Prediction Platforms and Mathematical Modeling of Aggregation Dynamics
AI and Machine Learning for Predicting Aggregation-Prone Regions (APRs)
Protein misfolding and aggregation are fundamental pathological processes in numerous neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), frontotemporal lobar degeneration (FTLD), and amyotrophic lateral sclerosis (ALS) [40] [17]. The accumulation of aggregates formed by pathological proteins such as amyloid-β, tau, α-synuclein, and TAR DNA-binding protein 43 (TDP-43) characterizes these devastating conditions [40]. These protein aggregates, particularly those rich in β-sheet structures, exhibit prion-like propagation, facilitating their spread throughout neural networks and ultimately inducing neurodegeneration [40] [17].
Within this context, the identification of aggregation-prone regions (APRs)—short sequence segments that initiate and drive the aggregation process—has emerged as a critical research focus. Traditionally, experimental methods to profile protein aggregation have been costly, labor-intensive, and time-consuming, creating a significant bottleneck in both basic research and therapeutic development [41]. The emergence of artificial intelligence (AI) and machine learning (ML) has revolutionized this field, providing researchers with powerful computational tools to predict APRs with unprecedented speed and accuracy. These advances are enhancing our fundamental understanding of protein aggregation and opening new avenues for therapeutic intervention in neurodegenerative diseases.
Fundamental Concepts and Biological Basis
APRs are typically short sequence stretches (usually 5-15 amino acids) that possess inherent tendencies to form stable, intermolecular β-sheet structures [17]. The aggregation process often begins with the formation of β-strand seed structures that act as nucleation sites for further aggregation [17]. Under pathological conditions such as gene mutations, abnormal post-translational modifications, or exposure to environmental stressors, the intramolecular and intermolecular interactions of native proteins are disrupted [40]. These changes facilitate β-sheet formation and enhance protein-protein interactions, leading to aggregation [40].
The resulting protein aggregates, rich in β-sheets, can act as templates to form de novo fibrils in a self-propagating manner [40]. This prion-like behavior enables the cell-to-cell transmission of pathological protein species, contributing to the progressive nature of neurodegenerative diseases [40] [17]. Notably, soluble oligomeric species formed during early aggregation stages are now regarded as primary drivers of neurotoxicity, characterized by their amorphous structures and exposed hydrophobic regions that render them highly reactive and hazardous to neuronal function [17].
Evolution of Computational Approaches
Early computational methods for APR prediction primarily relied on identifying sequence-based physicochemical properties associated with aggregation, such as hydrophobicity, charge, and secondary structure propensity [42]. These methods employed relatively simple algorithms like linear regression and support vector machines.
The field has since evolved dramatically with the incorporation of deep learning architectures and the integration of diverse data types. Modern approaches now leverage:
- Protein Language Models (e.g., ESM2) that learn evolutionary patterns from millions of protein sequences
- Structural Information from predictive tools like AlphaFold2
- Graph-Based Representations that capture residue-level interactions within protein structures
- Multi-Modal Learning frameworks that combine sequence, evolutionary, physicochemical, and structural information [41] [43]
This evolution has transformed APR prediction from a simple sequence-based scoring task to a sophisticated multi-dimensional analysis capable of capturing the complex biophysical determinants of aggregation.
State-of-the-Art AI Frameworks and Methodologies
AggNet: A Deep Learning Framework Integrating Multiple Data Modalities
AggNet represents a significant advancement in APR prediction, combining a protein language model (ESM2) with structural insights from AlphaFold2 to discriminate amyloid and non-amyloid peptides and identify APRs in diverse proteins [41]. The framework utilizes physicochemical, evolutionary, and structural information to achieve state-of-the-art performance.
Key Architectural Components:
- ESM2 Protein Language Model: Processes amino acid sequences to extract evolutionary information and sequence patterns correlated with aggregation
- AlphaFold2-Derived Structural Features: Incorporates predicted structural attributes that influence aggregation propensity
- Multi-Layer Deep Learning Classifier: Integrates diverse feature sets to generate final predictions on amyloid formation and APR localization
Benchmark comparisons demonstrate that AggNet outperforms existing methods and maintains stable predictive performance across proteins with different secondary structures [41]. Feature analysis and visualizations confirm that the model effectively captures relevant physicochemical properties, thereby offering enhanced interpretability beyond mere prediction [41].
Table 1: Performance Metrics of State-of-the-Art APR Prediction Tools
| Model Name | Architecture | Key Features | Reported Performance | Key Advantages |
|---|---|---|---|---|
| AggNet [41] | Deep Learning (ESM2 + AlphaFold2) | Physicochemical, evolutionary, structural | Outperforms existing methods (SOTA) | Stable across different secondary structures; Enhanced interpretability |
| GCN Model [43] | Graph Convolutional Network | Structural topology, residue interactions | R² = 0.9849, MAE = 0.0381 | Captures spatial relationships in protein structures |
| RibbonFold [44] | AI with physical constraints | Ribbon-like amyloid characteristics | Outperforms AlphaFold2 on amyloids | Specialized for amyloid fibrils; Reveals polymorph transitions |
Graph Convolutional Networks (GCNs) for Structure-Based Prediction
A recent study has constructed a Graph Convolutional Network (GCN) to predict PA scores using an expanded and refined dataset from the Protein Data Bank (PDB) and AlphaFold2.0 [43]. This approach explicitly models proteins as graphs where nodes represent amino acid residues and edges represent spatial interactions or chemical contacts between them.
Methodological Workflow:
- Data Curation: Systematically separate multi-polypeptide chains within PDB data into single polypeptide chains, removing redundancy
- Feature Extraction: Employ AGGRESCAN3D 2.0 to calculate PA propensity as ground truth labels
- Graph Construction: Represent protein structures as graphs with nodes (residues) and edges (spatial interactions)
- Model Training: Train GCN to learn mapping between graph representations and aggregation propensities
This GCN model achieves a remarkable coefficient of determination (R²) score of 0.9849 and a low mean absolute error (MAE) of 0.0381 [43]. The structural granularity of GCNs enables identification of specific residue interactions that contribute to aggregation propensity, moving beyond sequence-based patterns alone.
RibbonFold: Specialized Prediction of Amyloid Structures
RibbonFold addresses a critical gap in general protein structure prediction by specifically targeting the complex and variable structures of incorrectly folded proteins, particularly amyloids [44]. Unlike tools such as AlphaFold2 or AlphaFold3, which are trained on well-behaved, globular proteins, RibbonFold incorporates physical constraints suited to capture the ribbonlike characteristics of amyloid fibrils.
Innovative Aspects:
- Physical Constraints: Incorporates understanding of the energy landscape of amyloid fibrils
- Structural Polymorphism Prediction: Capable of predicting multiple stable forms (polymorphs) of amyloid fibrils
- Temporal Evolution: Suggests fibrils may begin in one structural form but shift into more insoluble configurations over time, explaining disease progression
RibbonFold's specialized approach reveals previously overlooked nuances in how amyloids form and evolve in the body, providing insights particularly relevant to the late onset of symptoms in neurodegenerative diseases [44].
Experimental Protocols and Methodologies
AggNet Implementation Workflow
Data Preparation and Feature Extraction
- Sequence Database Curation: Collect comprehensive datasets of amyloid and non-amyloid peptides from publicly available repositories
- Evolutionary Feature Extraction: Process sequences through ESM2 to generate embeddings capturing evolutionary constraints
- Structural Feature Generation: Obtain 3D structural predictions using AlphaFold2 for target sequences
- Physicochemical Descriptor Calculation: Compute hydrophobicity, charge, secondary structure propensity, and solvent accessibility features
Model Training and Validation
- Architecture Configuration: Implement a multi-branch neural network to process different feature types
- Cross-Validation: Employ k-fold cross-validation to ensure robustness across different protein families
- Benchmarking: Compare performance against established methods using standardized metrics
- Interpretability Analysis: Apply visualization techniques to identify features most influential for predictions
The resulting model has demonstrated practical utility in real-world applications, including a case study on MEDI1912 that confirmed AggNet's ability to guide mutation strategies for aggregation mitigation in protein therapeutics [41].
Active Learning Framework for Efficient Discovery
The GCN approach incorporated an active learning process to enhance the efficiency of identifying proteins with high aggregation propensity [43]. This methodology is particularly valuable for screening large protein datasets or designing aggregation-resistant biotherapeutics.
Active Learning Cycle:
- Initial Model Training: Train initial GCN model on available labeled data
- Uncertainty Sampling: Deploy model to predict on unlabeled data and select instances where model confidence is lowest
- Targeted Experimental Validation: Perform focused experimental testing on selected high-value targets
- Model Retraining: Incorporate new labeled data to improve model performance iteratively
This active learning approach achieved an MAE of 0.0291 in expected improvement, surpassing other methods, and identified 99% of the target proteins by exploring merely 29% of the entire search space [43]. This dramatically reduces the experimental burden required for comprehensive aggregation profiling.
Diagram 1: Comprehensive workflow for AI-driven APR prediction, integrating multiple data modalities and validation steps.
Implementing AI approaches for APR prediction requires access to specific computational resources, datasets, and software tools. The following table summarizes key components of the modern computational biologist's toolkit for aggregation research.
Table 2: Essential Research Resources for AI-Driven APR Prediction
| Resource Category | Specific Tools/Resources | Function/Purpose | Access Information |
|---|---|---|---|
| Protein Structure Databases | AlphaFold Protein Structure Database [45] | Provides >200 million protein structure predictions | Open access via https://alphafold.ebi.ac.uk/ |
| Specialized Prediction Tools | AggNet [41] | Predicts APRs using deep learning and protein language models | Source code: https://github.com/Hill-Wenka/AggNet |
| Specialized Prediction Tools | RibbonFold [44] | Predicts amyloid fibril structures specifically | Research implementation (contact authors) |
| Experimental Aggregation Data | Global Neurodegeneration Proteomics Consortium (GNPC) [7] | Large-scale proteomic data for neurodegenerative diseases | Available via AD Workbench (from July 2025) |
| Computational Frameworks | Graph Convolutional Networks (GCN) with Active Learning [43] | Structure-based aggregation prediction with efficient sampling | Custom implementation based on published architecture |
Applications in Neurodegenerative Disease Research and Therapeutics
Elucidating Disease Mechanisms
AI-driven APR prediction tools are providing unprecedented insights into the molecular mechanisms underlying neurodegenerative diseases. By identifying specific APRs in disease-associated proteins like Aβ, tau, and α-synuclein, researchers can pinpoint the initial molecular events that trigger the pathogenic cascade [40] [17]. These tools have revealed how different structural polymorphs (strains) of the same protein can lead to distinct disease phenotypes and progression patterns, explaining some of the clinical heterogeneity observed in conditions like Alzheimer's and Parkinson's diseases [40].
The prediction of cross-seeding and trans-seeding interactions—where aggregates of one protein can initiate aggregation of another—represents another critical application [40]. These interactions contribute to the pathological complexity observed in many neurodegenerative disease brains, where multiple co-pathologies frequently coexist. Understanding these interactions at the molecular level is essential for developing comprehensive therapeutic strategies.
Drug Discovery and Therapeutic Design
AI-based APR prediction is accelerating drug discovery in multiple ways:
Aggregation-Resistant Biotherapeutics
- Optimization of Therapeutic Proteins: AggNet has been successfully applied to guide mutations that reduce aggregation in therapeutic antibodies like MEDI1912, streamlining the development of more stable and effective biopharmaceuticals [41]
Small-Molecule Drug Development
- Structure-Based Inhibitor Design: Accurate APR and fibril structure predictions enable rational design of small molecules that specifically target and disrupt aggregation pathways [44]
- Strain-Specific Therapeutics: RibbonFold's ability to predict structural polymorphs facilitates development of strain-specific therapeutics that could target the most pathogenic aggregate forms [44]
Novel Target Identification
- tRNA Processing Pathways: Recent research has revealed unexpected connections between protein aggregation and disrupted tRNA processing in genetic forms of neurodegeneration [46]
- Cellular Clearance Mechanisms: APR predictions combined with proteomic analyses can identify novel components of cellular quality control systems that might be enhanced therapeutically
Future Directions and Challenges
Technical Advancements
The field of AI-driven APR prediction continues to evolve rapidly, with several promising directions emerging:
Integration with Multi-Omics Data Future frameworks will likely incorporate proteomic, transcriptomic, and genomic data to contextualize aggregation propensity within broader cellular pathways. Initiatives like the Global Neurodegeneration Proteomics Consortium (GNPC), which has established one of the world's largest harmonized proteomic datasets, will facilitate this integration [7]. The GNPC includes approximately 250 million unique protein measurements from over 35,000 biofluid samples, providing unprecedented resources for validation and discovery [7].
Temporal Prediction of Aggregation Current methods primarily predict static aggregation propensity, but the dynamic evolution of aggregates over time represents a critical frontier. RibbonFold's suggestion that fibrils may shift into more insoluble configurations over time points toward the need for models that can simulate temporal progression [44].
Enhanced Interpretability While current models like AggNet already offer some interpretability, future developments will focus on more explicitly linking specific sequence and structural features to aggregation mechanisms, providing deeper mechanistic insights rather than mere predictions.
Clinical Translation Challenges
Despite rapid technical progress, significant challenges remain in translating these computational advances into clinical impact:
Biological Complexity The sheer complexity of protein aggregation in living systems—influenced by cellular environment, post-translational modifications, oxidative stress, and molecular chaperones—presents ongoing challenges for accurate prediction [17]. Models must increasingly account for these contextual factors.
Validation in Heterogeneous Diseases Neurodegenerative diseases exhibit substantial heterogeneity in their pathological manifestations. Validating computational predictions across diverse patient populations and disease subtypes requires access to large, well-characterized tissue and biofluid collections [7].
Therapeutic Delivery Even with accurate APR predictions, delivering therapeutics effectively across the blood-brain barrier and targeting specific neuronal populations remains a formidable challenge that computational approaches alone cannot solve.
AI and machine learning have transformed our ability to identify and characterize aggregation-prone regions in proteins, providing powerful tools to unravel the complex mechanisms underlying neurodegenerative diseases. Frameworks like AggNet, GCNs, and RibbonFold represent significant advances over earlier methods, leveraging deep learning, structural information, and specialized architectures to achieve unprecedented predictive accuracy.
These computational approaches are not merely theoretical exercises but are already delivering practical benefits in basic research, target validation, and therapeutic design. As these tools continue to evolve—integrating diverse data types, improving interpretability, and capturing dynamic processes—they promise to accelerate the development of urgently needed therapies for neurodegenerative conditions. The integration of robust computational prediction with experimental validation creates a virtuous cycle of discovery, moving us closer to effective interventions for Alzheimer's disease, Parkinson's disease, and other protein aggregation disorders.
Coarse-Grained Molecular Dynamics Simulations for Evaluating Solvent-Accessible Surface Area
Coarse-grained molecular dynamics (CGMD) simulations have emerged as a powerful computational technique for studying biological processes that occur over spatial and temporal scales inaccessible to all-atom models. In the context of neurodegenerative diseases, where protein misfolding and aggregation play central pathological roles, CGMD provides a unique opportunity to investigate the long-timescale behavior of amyloidogenic proteins such as Aβ, tau, and α-synuclein [47] [11]. Within these simulations, the accurate estimation of solvent-accessible surface area (SASA) serves as a critical computational metric for quantifying solvation effects, protein stability, and aggregation propensity [48].
SASA represents the surface area of a biomolecule that is accessible to a solvent probe and serves as a fundamental parameter in implicit solvation models. For CGMD simulations of protein misfolding, SASA provides essential insights into hydrophobic burial during aggregation, interaction interfaces in oligomer formation, and solvation free energy contributions to protein stability [48]. This technical guide examines the integration of rapid SASA estimators with CGMD simulations, with specific application to the investigation of protein misfolding and aggregation mechanisms in neurodegenerative diseases.
Theoretical Foundations of Coarse-Grained Molecular Dynamics
Fundamental Principles of CGMD
Coarse-grained molecular dynamics is a computational approach that reduces system complexity by grouping multiple atoms into single interaction sites, known as beads. This systematic reduction of degrees of freedom enables simulations of larger systems and longer timescales compared to all-atom simulations [49] [50]. In CGMD, the continuum-level energy is derived from an ensemble average over atomic motions where atomic positions are constrained to produce the proper coarse-scale field [49]. This approach retains the thermodynamic average effect of fine-scale quantities not included in the coarse-scale motion.
The equations of motion in CGMD resemble those in finite element analysis but do not rely on continuum elastic theory assumptions. The method recovers full molecular dynamics in regions where mesh nodes coincide with atomic sites and transitions seamlessly to continuum elastic theory in the macroscopic limit [49]. A key advantage is that CGMD reproduces molecular dynamics behavior in atomic regions while enabling simulation of much larger regions through coarse-graining, making it particularly suitable for studying mesoscopic phenomena such as initial protein aggregation events.
CGMD Force Fields and Parametrization
Coarse-grained force fields describe effective interactions between beads, with functional forms that can range from simple analytical potentials to complicated tabulated functions [51]. These models are designed to reproduce specific properties of reference systems, which may include all-atom simulations or experimental data. Parameterization methods include:
- Free energy conservation approaches such as the simplex method
- Structure-based coarse-graining that conserves distributions like radial distribution functions using iterative Boltzmann inversion or inverse Monte Carlo
- Force matching methods that conserve forces [51]
The MARTINI force field represents one of the most widely used coarse-grained parameter sets for biomolecular systems, allowing for the construction of proteins and membranes [51]. Other approaches include the PLUM model, a solvent-free protein-membrane model derived from structure-based coarse-graining [51]. Importantly, coarse-grained potentials are state-dependent (varying with temperature, density, etc.) and generally require re-parametrization for different systems and simulation conditions.
Table 1: Comparison of Coarse-Grained Modeling Approaches
| Method | Mapping Resolution | Typical Applications | Time Step | Key Features |
|---|---|---|---|---|
| MARTINI | 3-5 heavy atoms per bead | Membranes, proteins, lipids | 20-40 fs | Balanced detail and efficiency |
| Cα-only models | 1 bead per amino acid | Protein folding, dynamics | 10-50 fs | Backbone-focused, simplified |
| Shape-based CG | Variable based on molecular shape | Large assemblies | 20-50 fs | Structure-based parametrization |
| Ultra-CG | Entire domains as single beads | Large conformational changes | >100 fs | Maximum simplification |
SASA Calculation in Coarse-Grained Simulations
Theoretical Significance of SASA
The solvent-accessible surface area serves as a fundamental geometric descriptor in molecular simulations, with particular importance in coarse-grained frameworks where atomic detail is sacrificed for computational efficiency. SASA models enable estimation of transfer free energies associated with biophysical processes such as protein folding, binding, and aggregation [48]. In the context of implicit solvent models, SASA provides a computationally efficient means to account for solvation effects without explicit water molecules, significantly accelerating simulations.
For protein misfolding research, SASA offers crucial insights into several aggregation-related phenomena:
- Hydrophobic exposure: Increasing SASA of nonpolar residues drives aggregation to minimize solvent contacts
- Oligomer surface characteristics: SASA patterns distinguish toxic oligomers from benign aggregates
- Interface formation: Reduced SASA at protein-protein interfaces indicates stable oligomerization
- Solvation energies: SASA-dependent terms estimate solvation free energy contributions to aggregation kinetics
SASA Calculation Methods for CG Structures
Computing SASA for coarse-grained structures presents unique challenges due to the reduced representation of molecular geometry. The PCASA (Protein-Cα Solvent Accessibilities) method represents a specialized approach designed specifically for Cα-only protein structures [48]. This method employs Bayesian linear regression to estimate residue-wise SASAs based solely on Cα coordinates, demonstrating superior performance compared to previous methods like POPS-R, particularly for unfolded protein conformations relevant to misfolding studies.
Alternative SASA computation methods include:
- Numerical integration: Using spherical probes with approximate surface algorithms
- Analytical methods: Applying mathematical approximations for simple geometric shapes
- Machine learning approaches: Training on all-atom simulations to predict SASA from CG coordinates
The integration of rapid SASA estimators like PCASA with CGMD simulations enables efficient inclusion of SASA-based solvent free energy estimations in protein folding and aggregation studies [48]. This combination allows researchers to maintain reasonable computational cost while incorporating essential solvation effects in their models.
Table 2: SASA Calculation Methods for Coarse-Grained Structures
| Method | Representation | Hydration Treatment | Computational Speed | Accuracy |
|---|---|---|---|---|
| PCASA | Cα-only | Implicit | Very Fast | High for unfolded states |
| Fast-SAXS | Coarse-grained residue level | Explicit water placement | Fast | Moderate |
| CRYSOL | Atomic | Implicit water layer | Moderate | High |
| FoXS | Atomic or coarse-grained | Implicit based on surface accessibility | Moderate to Fast | High |
| AquaSAXS | Atomic | AquaSol solvent density map | Moderate | High |
Integration of CGMD and SASA for Protein Misfolding Research
Protein Misfolding and Neurodegenerative Diseases
Neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), dementia with Lewy bodies (DLB), and others are characterized by the misfolding and aggregation of specific proteins [47] [11]. In AD, the amyloid-β (Aβ) peptide and tau protein misfold and form toxic aggregates, while in PD, α-synuclein aggregates represent the pathological hallmark [11]. The conversion of native proteins into amyloid aggregates involves a complex multistep process that proceeds through various intermediate states, including misfolded protein oligomers that are increasingly recognized as the primary cytotoxic agents in many neurodegenerative conditions [47].
The molecular mechanisms underlying protein misfolding and aggregation are difficult to investigate experimentally due to the transient nature of aggregation intermediates and the limited temporal and spatial resolution of experimental techniques. Molecular dynamics simulations, particularly CGMD, fill this gap by providing an atomic-level perspective on the step-by-step process of protein misfolding and aggregation [52]. When combined with experimental analyses, CGMD offers detailed insights into the molecular events driving neurodegenerative pathogenesis.
SASA as a Marker for Aggregation-Prone States
In CGMD simulations of protein misfolding, SASA serves as a valuable marker for identifying aggregation-prone states and intermediates. During the aggregation process, proteins typically undergo structural changes that expose hydrophobic regions normally buried in the native state, increasing their SASA and driving association with other misfolded proteins [47] [53]. The subsequent burial of these hydrophobic surfaces through protein-protein interactions then reduces SASA, providing a clear thermodynamic driving force for aggregation.
Research indicates that specific oligomeric species of Aβ and α-synuclein exhibit distinct structural characteristics and cytotoxic effects [47]. For Aβ, various oligomeric forms have been described, including spherical and chain-like protofibrils, paranuclei, pentamers, globulomers, amylospheroids, and Aβ-derived diffusible ligands (ADDLs) [47]. Similarly, for α-synuclein, type A, type A, type B, and type B oligomers have been identified [47]. Monitoring SASA patterns throughout oligomer formation and growth in CGMD simulations can help identify which species expose critical hydrophobic patches correlated with cellular toxicity.
Diagram 1: SASA Dynamics in Protein Aggregation Pathway. This workflow illustrates how SASA changes during protein misfolding and aggregation, with hydrophobic exposure driving initial association and subsequent burial stabilizing aggregates.
Computational Protocols and Methodologies
CGMD Simulation Setup for Misfolding Studies
Establishing a CGMD simulation for protein misfolding research requires careful consideration of several parameters:
System Preparation:
- Select appropriate coarse-grained mapping resolution (Cα-only, MARTINI, etc.)
- Generate initial protein structures (monomeric or oligomeric)
- Solvate the system using implicit or explicit coarse-grained solvent
- Incorporate ions to achieve physiological ionic strength
Force Field Selection:
- Choose force fields specific to amyloid-forming proteins when available
- Validate non-bonded interaction parameters for aggregation behavior
- Implement SASA-dependent energy terms if using implicit solvation
Simulation Parameters:
- Utilize integration time steps of 10-50 fs, significantly larger than atomistic simulations
- Implement temperature control through Langevin dynamics or Nosé-Hoover thermostats
- Apply periodic boundary conditions with appropriate long-range interaction treatments
- Conduct energy minimization prior to production dynamics
SASA Integration and Analysis Protocols
The integration of SASA calculations with CGMD simulations can be implemented through various approaches:
On-the-fly SASA Calculation:
- Implement PCASA or similar rapid estimators at each dynamics step
- Incorporate SASA-dependent energy terms for implicit solvation
- Calculate per-residue SASA values for aggregation analysis
Trajectory Analysis:
- Compute SASA time series for specific regions or residues
- Identify correlated SASA changes among multiple molecules
- Calculate hydrophobic exposure metrics for aggregation propensity
Analysis of Aggregation States:
- Cluster structures based on SASA patterns and oligomeric states
- Calculate free energy landscapes as functions of SASA and other order parameters
- Identify critical SASA thresholds associated with oligomer formation
Diagram 2: CGMD-SASA Integration Workflow. This protocol outlines the key steps in integrating SASA calculations with CGMD simulations for protein misfolding studies.
Research Reagent Solutions
Table 3: Essential Computational Tools for CGMD-SASA Research
| Tool Category | Specific Software/Package | Primary Function | Application in Misfolding Studies |
|---|---|---|---|
| Simulation Engines | GROMACS [51] | Molecular dynamics simulation | Running CGMD simulations with various force fields |
| Coarse-Graining Tools | VOTCA [51] | Systematic coarse-graining | Parameterizing CG models for specific proteins |
| SASA Calculators | PCASA [48] | Solvent-accessible surface area estimation | Rapid SASA calculation from Cα coordinates |
| Analysis Suites | WESTPA [50] | Weighted ensemble sampling | Enhanced sampling for rare aggregation events |
| Force Fields | MARTINI [51] | Coarse-grained parameters | Simulating protein-lipid interactions in neurodegeneration |
| Visualization | Chimera [54] | Molecular visualization | Analyzing SASA patterns and aggregation interfaces |
Applications to Neurodegenerative Disease Mechanisms
Investigating Aβ and Tau Aggregation in Alzheimer's Disease
CGMD simulations combined with SASA analysis have provided molecular insights into the aggregation mechanisms of Alzheimer's-related proteins. For Aβ, simulations have revealed how hydrophobic exposure drives the initial association of monomers and the formation of various oligomeric species with distinct toxic properties [47] [53]. SASA analysis helps identify critical hydrophobic patches that become exposed during partial unfolding and how their burial correlates with stabilization of oligomeric interfaces.
In the case of tau protein, CGMD simulations have examined how post-translational modifications affect protein structure and aggregation propensity. SASA calculations quantify the exposure of specific domains that promote tau-tau interactions or mediate binding to microtubules. The combination of CGMD and SASA has been particularly valuable in studying the polymorphic nature of amyloid fibrils, which adopt different structures in different patients and disease contexts [47].
α-Synuclein Oligomerization in Parkinson's Disease
The aggregation of α-synuclein in Parkinson's disease follows a complex pathway involving multiple oligomeric intermediates. CGMD simulations have identified how environmental factors such as oxidative stress, lipid interactions, and molecular crowding influence α-synuclein conformation and oligomerization kinetics [47] [11]. SASA analysis provides quantitative metrics for how specific mutations (e.g., A53T, A30P, E46K) associated with familial PD alter hydrophobic exposure and aggregation propensity.
Recent research has identified distinct α-synuclein oligomer types (A, A, B, and B) with different structural characteristics and cytotoxic effects [47]. CGMD simulations with SASA monitoring can reconstruct the formation pathways of these oligomers and identify the key structural features that distinguish them. This approach offers the potential to design therapeutic strategies that selectively target the most toxic oligomeric species while sparing functional forms of the protein.
Advanced Technical Considerations
Validation Against Experimental Data
Validating CGMD simulations and SASA predictions against experimental data is essential for establishing their biological relevance. Several experimental techniques provide complementary data for validation:
Small-Angle X-ray Scattering (SAXS) provides low-resolution structural information about proteins in solution and can be used to validate overall dimensions and oligomeric states predicted by simulations [54]. Computational methods such as FoXS and CRYSOL enable calculation of theoretical SAXS profiles from atomic models for direct comparison with experimental data [54].
Cryo-Electron Microscopy (cryo-EM) has revolutionized structural biology of amyloid fibrils, providing high-resolution structures of patient-derived fibrils [47]. These structures serve as essential references for validating simulation outcomes.
Nuclear Magnetic Resonance (NMR) spectroscopy offers information about protein dynamics and transient structures that can validate simulation-predicted conformational ensembles.
Enhanced Sampling Techniques
Protein aggregation involves rare events that occur on timescales challenging to access even with CGMD. Enhanced sampling techniques address this limitation:
Weighted Ensemble (WE) methods such as WESTPA run multiple trajectory replicas with periodic resampling based on progress coordinates, increasing the likelihood of observing rare events like oligomer formation [50].
Metadynamics accelerates sampling by adding bias potentials along selected collective variables, such as SASA or intermolecular contacts.
Temperature-based enhanced sampling techniques like simulated tempering improve conformational sampling by simulating at multiple temperatures.
These advanced computational approaches, combined with experimental validation, strengthen the biological insights derived from CGMD simulations of protein misfolding and aggregation.
The integration of coarse-grained molecular dynamics simulations with solvent-accessible surface area analysis represents a powerful methodology for investigating protein misfolding and aggregation in neurodegenerative diseases. This approach provides the temporal and spatial resolution necessary to study molecular mechanisms that are difficult to capture experimentally, while maintaining computational efficiency through appropriate coarse-graining. As force fields continue to improve and SASA estimators become more sophisticated, CGMD simulations will play an increasingly important role in elucidating the structural principles of protein misfolding, identifying toxic oligomeric species, and supporting therapeutic development for devastating neurodegenerative conditions.
Genetic Algorithms and Reinforcement Learning for De Novo Peptide Design
The accumulation of misfolded proteins is a hallmark of numerous neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease [10] [12] [11]. In these conditions, specific proteins such as β-amyloid (Aβ), tau, α-synuclein, and huntingtin undergo conformational changes that lead to aggregation into toxic oligomers and insoluble fibrils, ultimately resulting in neuronal dysfunction and cell death [10] [11]. The central challenge in developing therapeutic interventions lies in designing molecules that can specifically target and modulate these aggregation processes.
The emergence of artificial intelligence (AI) has revolutionized the computational design of peptides, offering powerful tools to explore the vast combinatorial sequence space far beyond the capabilities of traditional trial-and-error experimental approaches [55]. Among these AI strategies, genetic algorithms (GAs) and reinforcement learning (RL) have shown exceptional promise for the de novo design of peptides with tailored aggregation propensities and binding specificities [55] [56]. These methods enable researchers to navigate the immense design space of potential peptide sequences systematically, generating candidates that can potentially inhibit the misfolding and aggregation pathways central to neurodegenerative diseases.
Core Computational Methodologies
Genetic Algorithms for Peptide Optimization
Genetic algorithms are evolutionary computation techniques inspired by natural selection that iteratively evolve populations of candidate solutions toward optimal designs [55] [57]. In the context of peptide design, GAs operate through a cyclical process of selection, crossover, and mutation to progressively improve peptide sequences based on defined fitness criteria.
A recent groundbreaking study demonstrated the application of GAs for designing decapeptides with tunable aggregation propensities [55]. The researchers started with 1,000 randomly generated initial sequences and implemented a mutation rate of 1%, meaning each residue had a 1% probability of being replaced by another amino acid during each generation. Through 500 iterations, the algorithm successfully evolved peptides from an average aggregation propensity (AP) of 1.76 to 2.15, effectively transforming low aggregation propensity peptides (LAPPs) into high aggregation propensity peptides (HAPPs) [55]. The workflow successfully identified sequences like WFLFFFLFFW (AP = 2.24) that formed large aggregates in molecular dynamics simulations, while control sequences like VMDNAELDAQ (AP = 1.14) remained uniformly distributed in solution [55].
Table 1: Key Genetic Algorithm Applications in Peptide Design
| Application Domain | Algorithm Features | Key Outcomes | Citation |
|---|---|---|---|
| Aggregation Propensity Tuning | Population: 1000 sequences, 1% mutation rate, 500 generations | Increased average AP from 1.76 to 2.15; Designed WFLFFFLFFW (AP=2.24) | [55] |
| Antimicrobial Peptide Design | Codon-based representation (CB-GA) with rough set theory | Customized AMPs active against S. epidermidis with improved synthesis properties | [57] |
| Heterotrimeric Collagen Design | GRACE algorithm with SCEPTTr1.2 scoring function | Designed peptides self-assembled into target triple helices with minimum specificity of 13.5°C | [58] |
| Chemical Space Exploration | Peptide Design GA (PDGA) using MXFP fingerprint space | Generated high-similarity analogues of bioactive peptides with diverse topologies | [59] |
Reinforcement Learning for Targeted Design
Reinforcement learning approaches, particularly those incorporating Monte Carlo Tree Search (MCTS), have demonstrated remarkable success in target-specific peptide design by framing the sequence generation problem as a series of decisions that maximize a reward function [55] [56].
The CYCBUILDER framework exemplifies this methodology for designing cyclic peptide binders, which are particularly valuable for targeting protein-protein interactions implicated in neurodegenerative pathways [56]. This RL-based system assembles peptide fragments and performs efficient cyclization via head-to-tail amide or disulfide bonds, using MCTS to guide fragment selection, peptide growth, and structure refinement. When applied to generate TNFα inhibitors, CYCBUILDER outperformed existing methods (AfCycDesign and Anchor Extension) in binding energy, structural diversity, and efficiency [56]. Experimental validation confirmed that four of nine designed peptides exhibited potent binding and cellular activity, demonstrating the practical efficacy of this approach [56].
Another innovative integration of reinforcement learning with deep learning demonstrated the ability to make targeted optimizations of peptide sequences while preserving desired functional features [55]. By combining a Transformer-based prediction model with MCTS, researchers achieved targeted optimization of peptide sequences, enabling the transformation of a non-aggregating decapeptide into a highly aggregative variant by replacing only two residues [55].
Integrated AI-MD Workflows
The most effective peptide design strategies combine AI-based generation with physics-based simulation methods in hierarchical workflows. One such approach integrates a Gated Recurrent Unit-based Variational Autoencoder (GRU-VAE) with Rosetta FlexPepDock and molecular dynamics (MD) simulations [60]. This methodology first uses the VAE with Metropolis-Hasting sampling to reduce the sequence search space from millions to hundreds of candidates, then employs physics-based methods for binding assessment, and finally uses MD simulations with MM/GBSA binding energy calculations to select the most promising candidates [60].
When applied to design peptide inhibitors targeting β-catenin, this integrated approach produced twelve candidates, six of which exhibited improved binding affinity compared to the parent peptide [60]. The most successful C-terminal peptide bound β-catenin with an IC₅₀ of 0.010 ± 0.06 μM, representing a 15-fold improvement over the original peptide [60]. Similarly, for NF-κB essential modulator (NEMO), two of four tested peptides showed substantially enhanced binding compared to the parent peptide [60].
Table 2: Performance Metrics of AI-Designed Peptides
| Target | AI Method | Experimental Validation Results | Citation |
|---|---|---|---|
| β-catenin | GRU-VAE + FlexPepDock + MD | 6/12 peptides showed improved binding; Best candidate: 15-fold improvement (IC₅₀ = 0.010 ± 0.06 μM) | [60] |
| NF-κB essential modulator | GRU-VAE + FlexPepDock + MD | 2/4 peptides showed substantially enhanced binding | [60] |
| TNFα | CYC_BUILDER (RL with MCTS) | 4/9 designed peptides showed potent binding and cellular activity | [56] |
| S. epidermidis | GA + Rough Set Theory | Active peptides with improved aggregation propensity for synthesis | [57] |
Experimental Protocols and Methodologies
Aggregation Propensity Assessment Protocol
The accurate evaluation of peptide aggregation propensity is fundamental to designing therapeutics for neurodegenerative diseases. The following protocol has been successfully employed for assessing decapeptides [55]:
System Preparation:
- Construct a simulation box with peptides randomly distributed in aqueous solution.
- Apply a minimum inter-peptide distance constraint of 0.4 nm to prevent pre-aggregation.
- Use the Martini force field for coarse-grained molecular dynamics (CGMD) simulations.
Simulation Parameters:
- Run CGMD simulations for 125 ns, which has been demonstrated sufficient to identify differences in aggregation propensity between HAPPs and LAPPs.
- Maintain constant temperature and pressure conditions appropriate for the system.
Aggregation Propensity Calculation:
- Calculate solvent-accessible surface area (SASA) at the beginning (SASAinitial) and end (SASAfinal) of the simulation.
- Compute aggregation propensity (AP) using the formula: AP = SASAinitial / SASAfinal.
- Apply a threshold of AP > 1.5 to classify peptides as HAPPs (high aggregation propensity peptides), while peptides with AP < 1.5 are classified as LAPPs (low aggregation propensity peptides).
AI Model Integration:
- Train a Transformer-based deep learning model with self-attention mechanisms using CGMD simulation data.
- Use the model as a rapid proxy for CGMD simulations, reducing assessment time from hours to milliseconds.
Genetic Algorithm Implementation for Peptide Design
This protocol outlines the codon-based genetic algorithm approach that has demonstrated success in designing antimicrobial peptides with relevance to aggregation prevention [57]:
Initialization:
- Generate an initial population of peptide sequences (typically 100-1000 individuals).
- Convert peptide sequences to codon-representations to increase sequence diversity through aggressive mutations.
Fitness Evaluation:
- Apply transparent machine learning methods, such as rough set theory, to classify peptides according to desired activity.
- Define fitness functions based on multiple constraints, including target binding affinity, aggregation propensity, and synthetic feasibility.
- For neurodegenerative disease applications, include specific fitness penalties for sequences with amyloidogenic patterns.
Evolutionary Operations:
- Implement tournament selection to choose parent sequences based on fitness scores.
- Apply single-point or multi-point crossover with a probability of 0.6-0.8.
- Perform mutation operations on a codon basis, allowing substitutions, insertions, or deletions of single DNA bases with a probability of 0.01-0.05 per position.
Termination and Analysis:
- Run the algorithm for 100-500 generations or until convergence criteria are met.
- Analyze the evolutionary trajectory to identify sequence features correlated with improved fitness.
- Validate top-performing sequences through in silico simulations before experimental testing.
Visualization of Workflows and Signaling Pathways
Genetic Algorithm Peptide Design Workflow
Integrated AI-MD Peptide Design Pipeline
Protein Misfolding Pathways in Neurodegeneration
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Research Reagent Solutions for AI-Guided Peptide Design
| Reagent/Resource | Function | Application Context |
|---|---|---|
| Martini Force Field | Coarse-grained molecular dynamics parameters | Enables efficient simulation of peptide aggregation over relevant timescales (125 ns) [55] |
| Rosetta FlexPepDock | Flexible peptide-protein docking | Provides binding energy assessments for peptide-target interactions [60] |
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area | Calculates binding free energies from MD trajectories [60] |
| SCEPTTr 1.2 | Collagen triple helix stability prediction | Specialized scoring function for collagen-mimetic peptide design [58] |
| Codon-Based Representation | DNA-level sequence encoding | Enhances genetic algorithm diversity through reading frame manipulation [57] |
| Transformer Models with Self-Attention | Sequence-to-property prediction | Achieves high accuracy (94%+) in predicting aggregation propensity [55] |
| Rough Set Theory | Transparent machine learning classification | Provides interpretable decision boundaries for peptide activity [57] |
The integration of genetic algorithms and reinforcement learning with molecular modeling represents a paradigm shift in peptide design for neurodegenerative diseases. These computational approaches enable the systematic exploration of sequence spaces that are intractable through experimental methods alone, generating candidates with precise aggregation propensities and target specificities [55] [56] [60]. The successful experimental validation of AI-designed peptides against targets like β-catenin and TNFα demonstrates the practical potential of these methodologies [56] [60].
Future developments in this field will likely focus on improved multi-objective optimization strategies that simultaneously address aggregation propensity, membrane permeability, metabolic stability, and low immunogenicity – all critical factors for therapeutics targeting neurodegenerative conditions. Additionally, the incorporation of experimental data directly into the optimization loop through active learning approaches will further enhance the efficiency and success rates of computational design pipelines.
As our understanding of protein misfolding mechanisms in diseases like Alzheimer's and Parkinson's continues to evolve, these AI-driven peptide design methodologies offer promising avenues for developing targeted interventions that can disrupt the aggregation processes at the molecular level, potentially leading to effective treatments for these devastating conditions.
The study of protein aggregation is a cornerstone of neurodegenerative disease research, with mathematical models providing an essential framework for quantifying and predicting the kinetics of these complex processes. The histopathological presence of misfolded protein aggregates has been identified as primary evidence in multiple neurological diseases, including Alzheimer's disease, Parkinson's disease, and prion disorders [61]. Despite their biological importance, the precise mechanisms governing protein aggregation, propagation, and toxicity remain incompletely understood, creating a critical role for mathematical modeling approaches [61].
Ordinary differential equations (ODEs) have served as the fundamental mathematical language for describing aggregation kinetics since the earliest formalizations of these processes. These models are typically derived from mass action kinetics, which states that the rate of a reaction is proportional to the product of the concentrations of the reactants [62]. This review traces the historical development of ODE-based aggregation models, from their origins in classical enzymology to contemporary frameworks that capture the multi-step, multi-scale nature of protein aggregation in neurodegenerative diseases, providing researchers with both theoretical background and practical methodological guidance.
Historical Foundations: From Classical Enzymology to Early Aggregation Models
Michaelis-Menten Kinetics and the Briggs-Haldane Steady-State Approximation
The conceptual foundation for modeling biochemical kinetics was established nearly a century ago through the pioneering work of Michaelis and Menten (1913), later refined by Briggs and Haldane (1925) [62]. Their formulation described enzyme-catalyzed reactions using a system of ODEs based on mass action principles:
where E represents enzyme, S substrate, ES the enzyme-substrate complex, and P product. The Briggs-Haldane steady-state approximation assumed that the complex ES rapidly achieves a steady state, leading to the familiar Michaelis-Menten equation for reaction velocity. This established a critical precedent: complex biochemical processes could be approximated through simplified mathematical representations that capture essential dynamics while remaining tractable for analysis [62].
The Emergence of Protein Aggregation Models
The earliest mathematical models specifically addressing protein misfolding emerged from prion disease research. Eigen's pioneering work in the 1970s utilized ODEs to simulate autocatalysis mechanisms underlying prion replication, describing the conversion of normal prion protein (PrP^C) into its infectious counterpart (PrP^Sc) [61]. These initial models revealed limitations in simple heterodimer mechanisms, which could not replicate the long incubation periods characteristic of prion diseases.
This led to the development of the Nucleated Polymerization Model (NPM), which introduced several key concepts that would become fundamental to aggregation modeling:
- A critical threshold of protein concentration below which polymerization is negligible
- A lag time before observable polymerization begins
- The formation of stable nuclei that grow through incorporation of monomers
- Fragmentation processes that generate new nucleation sites [61]
Table 1: Historical Evolution of Key Aggregation Models
| Model | Time Period | Key Mathematical Features | Biological Interpretation |
|---|---|---|---|
| Michaelis-Menten | 1913-1925 | System of ODEs with steady-state approximation | Enzyme-substrate kinetics under well-mixed conditions |
| Heterodimer (Template-assisted) | 1970s | Simple autocatalytic ODE system | Prion conversion through direct protein-protein interaction |
| Nucleated Polymerization (NPM) | 1980s | ODE system with critical concentration threshold | Multi-step aggregation requiring stable nucleus formation |
| Smoluchowski Coagulation | 1990s-Present | Infinite ODE system describing aggregate size distribution | Population dynamics of aggregates of different sizes |
Fundamental Mathematical Frameworks for Aggregation Kinetics
Mass Action Kinetics and Deterministic Modeling Approaches
The majority of aggregation models are built upon the principle of mass action kinetics, which provides a deterministic framework for describing reaction rates in well-mixed systems [62]. This approach assumes continuous concentration variables and becomes valid when reactant molecule numbers exceed approximately 10²-10³, ensuring stochastic fluctuations become negligible relative to mean concentrations [62]. For neurodegenerative disease applications, where protein numbers typically satisfy this requirement, deterministic ODE models offer an effective balance between biological realism and computational tractability.
The general form of an ODE-based aggregation model follows:
where X represents the vector of chemical species concentrations, θ the kinetic parameters, and F the nonlinear function derived from mass action principles describing the interaction network.
The Smoluchowski Coagulation-Fragmentation Framework
The Smoluchowski coagulation equations provide a comprehensive mathematical framework for describing the time evolution of aggregate size distributions [63]. This approach considers the entire population of aggregates, with separate equations for each aggregate size:
where [i] denotes the concentration of aggregates containing i monomers, a{i,j} represents the coagulation rate constant between aggregates of sizes i and j, and f{i,j} represents the fragmentation rate constant for splitting an aggregate of size i+j into fragments of sizes i and j [63].
This infinite system of ODEs can be adapted to specific biological contexts through appropriate selection of coagulation and fragmentation kernels (a{i,j} and f{i,j}), which define the size-dependence of aggregation and fragmentation rates.
Diagram 1: Fundamental aggregation pathway showing nucleation, elongation, and fragmentation processes.
Application to Neurodegenerative Diseases: Specific Protein Aggregation Models
Amyloid-β and Tau Aggregation in Alzheimer's Disease
In Alzheimer's disease, mathematical models have been extensively applied to understand the aggregation kinetics of both amyloid-β (Aβ) and tau proteins. The amyloid cascade hypothesis posits that misfolding and aggregation of Aβ initiates a pathological cascade culminating in neurodegeneration [11]. Aβ aggregation follows a nucleation-dependent polymerization mechanism, where the rate-limiting step is formation of a stable nucleus, followed by rapid elongation through monomer addition.
Experimental studies have demonstrated that the Aβ42 isoform, though less abundant than Aβ40, exhibits significantly higher aggregation propensity and constitutes the primary component of amyloid plaques [9]. This observation can be captured mathematically through differential rate constants for nucleation and elongation processes between Aβ isoforms.
Tau protein aggregation follows distinct kinetics, characterized by intracellular neurofibrillary tangle formation. Models of tau aggregation must account for post-translational modifications (particularly phosphorylation) that significantly enhance aggregation propensity [9]. Additionally, the prion-like cell-to-cell spreading of tau pathology requires models that incorporate spatial propagation components alongside local aggregation kinetics.
α-Synuclein Aggregation in Parkinson's Disease
Parkinson's disease pathogenesis involves the multistep aggregation of α-synuclein, beginning with protein misfolding and progressing through oligomers, protofibrils, and ultimately insoluble fibrils that form Lewy bodies [11]. Mathematical models of α-synuclein aggregation must account for several unique features:
- The influence of oxidative stress on aggregation rates
- Membrane-mediated aggregation effects
- Strain-like polymorphism in aggregate structures
- Interactions with protein quality control systems
The synucleinopathy hypothesis suggests that α-synuclein aggregates induce neurotoxicity through multiple parallel mechanisms, including lysosomal impairment, mitochondrial dysfunction, and synaptic transmission disruption [11]. Comprehensive models therefore require coupling aggregation kinetics with cellular toxicity readouts.
Table 2: Experimentally Measured Kinetic Parameters for Neurodegenerative Disease-Related Protein Aggregation
| Protein | Nucleation Rate (M⁻¹s⁻¹) | Elongation Rate (M⁻¹s⁻¹) | Fragmentation Rate (s⁻¹) | Critical Concentration (μM) |
|---|---|---|---|---|
| Aβ42 | 10⁻³ - 10⁻¹ | 10⁴ - 10⁶ | 10⁻⁷ - 10⁻⁵ | 1 - 10 |
| α-Synuclein | 10⁻⁴ - 10⁻² | 10³ - 10⁵ | 10⁻⁸ - 10⁻⁶ | 5 - 20 |
| Tau | 10⁻⁵ - 10⁻³ | 10² - 10⁴ | 10⁻⁹ - 10⁻⁷ | 10 - 50 |
| TTR | 10⁻³ - 10⁻¹ | 10⁴ - 10⁵ | 10⁻⁷ - 10⁻⁵ | 2 - 15 |
Simplified Two-Equation Models for Practical Applications
While detailed aggregation models can involve numerous equations, simplified two-equation models have demonstrated remarkable utility for practical applications. These models reduce complexity by grouping all multi-mer species into a single variable, dramatically decreasing computational requirements while preserving essential dynamics [63].
A typical two-equation model for receptor clustering or protein aggregation takes the form:
where [M] represents monomer concentration, [C] represents cluster concentration, and kᵢ are kinetic parameters [63]. Such simplified models have successfully described diverse aggregation phenomena including GPVI or CR3 receptor clustering (2D) and albumin or platelet aggregation (3D) [63].
Experimental Methodologies for Parameter Estimation
In Vitro Aggregation Assays and Kinetic Analysis
Quantitative characterization of aggregation kinetics requires carefully controlled in vitro experiments. The following protocol outlines a standard approach for measuring protein aggregation kinetics:
Materials and Reagents:
- Purified recombinant protein (e.g., Aβ42, α-synuclein, tau)
- Aggregation buffer (typically PBS with optional stirring agents)
- Thioflavin T (ThT) or other fluorescent amyloid-binding dyes
- 96-well plates with non-binding surface
- Fluorescent plate reader with temperature control
Experimental Procedure:
- Prepare protein solution in denaturing conditions (e.g., 6M GdnHCl)
- Purify via size exclusion chromatography into aggregation buffer
- Add Thioflavin T to final concentration of 10-20 μM
- Aliquot protein-dye mixture into 96-well plate (100-200 μL per well)
- Seal plate to prevent evaporation and place in plate reader
- Measure fluorescence with excitation 440 nm, emission 480 nm
- Maintain constant temperature with continuous orbital shaking
- Collect data points every 5-10 minutes over 24-72 hours
Data Analysis:
- Normalize fluorescence readings to background (buffer + dye)
- Fit normalized data to appropriate kinetic model (typically sigmoidal)
- Extract key parameters: lag time, growth rate, and maximum amplitude
- Repeat at multiple protein concentrations to determine rate laws
The resulting sigmoidal curves characteristic of nucleated polymerization processes provide three key parameters: lag time (related to nucleation rate), growth rate (related to elongation rate), and maximum amplitude (related to total aggregate mass) [61].
Diagram 2: Experimental workflow for protein aggregation kinetics.
Parameter Estimation and Model Validation Techniques
Estimating kinetic parameters from experimental data represents a significant challenge in aggregation modeling. Nonlinear regression techniques, particularly maximum likelihood estimation and Bayesian approaches, are commonly employed. The development of the AmyloidFit software and related tools has partially automated this process, but careful statistical analysis remains essential [61].
Model validation should incorporate multiple experimental datasets, ideally including both macroscopic (e.g., ThT fluorescence) and microscopic (e.g., aggregate size distributions from microscopy) measurements. Techniques such as cross-validation, residual analysis, and comparison with alternative models help establish model credibility [64].
Table 3: Essential Research Reagents for Aggregation Kinetics Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Amyloid-Sensitive Dyes | Thioflavin T, Thioflavin S, Congo Red | Detection and quantification of amyloid structures | Varying specificity for different aggregate morphologies |
| Recombinant Proteins | Aβ1-42, α-synuclein, tau (various isoforms) | Provide controlled aggregation substrate | Purification method critically impacts aggregation propensity |
| Aggregation Modulators | Molecular chaperones (Hsp70), small molecule inhibitors | Mechanistic studies and therapeutic development | Specificity, potency, and mechanism of action |
| Analytical Standards | Size exclusion chromatography markers, pre-formed fibrils | Method calibration and seeding experiments | Reproducibility between batches is essential |
| Cell Culture Models | SH-SY5Y, primary neuronal cultures, iPSC-derived neurons | Cellular toxicity and propagation studies | Relevance to human pathophysiology |
Contemporary Extensions and Future Directions
Multi-Scale Modeling Frameworks
Contemporary research increasingly recognizes that protein aggregation in neurodegenerative diseases operates across multiple spatial and temporal scales. Modern modeling frameworks therefore integrate molecular-level aggregation kinetics with larger-scale processes including:
- Intercellular propagation through prion-like spreading mechanisms
- Spatial patterning of pathology through neural connectivity networks
- Protein homeostasis network interactions including ubiquitin-proteasome and autophagy-lysosome systems [11]
These multi-scale models typically employ hybrid approaches, combining ODE-based local aggregation kinetics with partial differential equations or agent-based methods for spatial propagation.
Therapeutic Applications and Drug Development
Mathematical models of aggregation kinetics have become invaluable tools in therapeutic development for neurodegenerative diseases. They enable quantitative prediction of therapeutic intervention effects, helping prioritize candidates for costly clinical trials [65]. Current therapeutic strategies targeting aggregation include:
- Stabilization of native protein structures (e.g., tafamidis for transthyretin amyloidosis)
- Inhibition of specific aggregation steps (nucleation, elongation, or secondary nucleation)
- Enhancement of cellular clearance pathways (autophagy, proteasomal degradation)
- Immunotherapeutic approaches that promote aggregate clearance [65] [11]
The successful development of tafamidis, which originated from basic research on transthyretin misfolding, demonstrates how mechanistic understanding of aggregation kinetics can translate into clinically effective therapies [65]. This small molecule drug stabilizes the tetrameric structure of transthyretin, preventing dissociation into aggregation-prone monomers and dramatically slowing disease progression in TTR amyloidosis [65].
Mathematical modeling of aggregation kinetics has evolved substantially from its origins in classical enzyme kinetics to sophisticated frameworks that capture the multi-step, multi-scale nature of protein aggregation in neurodegenerative diseases. ODE-based models remain fundamental tools for quantifying these processes, providing insights into mechanistic principles and enabling therapeutic development. As experimental techniques continue advancing, particularly in single-molecule imaging and structural biology, mathematical models will play an increasingly vital role in integrating diverse data types and predicting disease progression and treatment outcomes. The ongoing integration of mathematical modeling with experimental neuroscience promises to accelerate the development of effective therapies for these devastating disorders.
Integrating Structural Prediction Tools (AlphaFold) with Aggregation Risk Assessment
The integration of advanced structural prediction tools like AlphaFold with emerging methodologies for assessing protein aggregation risk represents a transformative approach in neurodegenerative disease research. This technical guide provides a comprehensive framework for leveraging AlphaFold2 (AF2) predictions to identify aggregation-prone regions, model the structural impact of genetic variants, and assess protein interaction interfaces. Within the broader context of protein misfolding and aggregation in neurodegenerative diseases, we detail experimental protocols for structural modeling, aggregation propensity analysis, and functional validation. By synthesizing current research and quantitative findings, this whitepaper equips researchers with methodologies to bridge computational structural biology with experimental aggregation risk assessment, potentially accelerating therapeutic development for conditions including Alzheimer's disease, Parkinson's disease, and related neurodegenerative disorders.
Neurodegenerative diseases constitute a major global health challenge characterized by the progressive loss of neuronal populations and associated cognitive and motor functions. A unifying pathological hallmark across these disorders is the aberrant misfolding and aggregation of specific proteins, which accumulate as toxic entities that drive neurodegeneration [11]. The major neurodegenerative conditions including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), frontotemporal lobar degeneration (FTLD), and amyotrophic lateral sclerosis (ALS) are each associated with the aggregation of particular proteins [40].
At the molecular level, the aggregation process involves a shift from native protein conformations to enriched β-sheet structures that assemble into oligomers, fibrils, and ultimately insoluble aggregates [11]. In AD, the key pathological proteins are amyloid-β (Aβ), which forms extracellular senile plaques, and hyperphosphorylated tau, which constitutes intracellular neurofibrillary tangles [40] [66]. PD and dementia with Lewy bodies are characterized by aggregates of α-synuclein within Lewy bodies and Lewy neurites [11]. These misfolded proteins can undergo prion-like propagation, with different structural conformations (strains) potentially accounting for disease heterogeneity [40].
The precise mechanisms of neurotoxicity remain incompletely understood but may include synaptic dysfunction, proteostasis disruption, mitochondrial impairment, and inflammatory responses [11] [67]. Understanding the structural basis of protein misfolding and aggregation is therefore critical for elucidating disease mechanisms and developing targeted therapeutic interventions.
AlphaFold2: Technical Foundation and Capabilities
AlphaFold2 represents a breakthrough in computational structural biology, providing the first computational method capable of predicting protein structures with atomic accuracy competitive with experimental determinations in the majority of cases [68]. The system employs a novel neural network architecture that incorporates physical and biological knowledge about protein structure, leveraging multi-sequence alignments (MSAs) into the deep learning algorithm design [68].
The network comprises two primary components: the Evoformer and the structure module. The Evoformer processes inputs through repeated layers of a novel neural network block that operates on both an MSA representation and a pair representation, enabling reasoning about spatial and evolutionary relationships between residues [68]. The structure module then introduces an explicit 3D structure through a series of transformations, starting from an initial state and progressively refining toward a highly accurate protein structure with precise atomic details [68].
Key Innovations and Performance
AlphaFold2 incorporates several groundbreaking technical innovations that enable its exceptional predictive accuracy:
- Novel output representation and loss function enabling end-to-end structure prediction
- Equivariant attention architecture respecting the geometric constraints of 3D space
- Iterative refinement through recycling with intermediate losses
- Masked MSA loss for joint training with structure
- Self-distillation from unlabeled protein sequences
- Integrated confidence self-estimates via predicted local-distance difference test (pLDDT) [68]
In the critical CASP14 assessment, AlphaFold2 demonstrated unprecedented accuracy with a median backbone accuracy of 0.96 Å r.m.s.d.95, dramatically outperforming competing methods which achieved 2.8 Å median accuracy [68]. This level of accuracy approaches experimental uncertainty, with the all-atom accuracy of 1.5 Å compared to 3.5 Å for the next best method [68].
Methodological Framework: Integrating AF2 with Aggregation Risk Assessment
The integration of AlphaFold2 predictions with aggregation risk assessment involves a multi-stage computational and experimental workflow. This systematic approach enables researchers to move from sequence-based identification of potential aggregation-prone regions to structural and functional validation.
Experimental Protocol 1: AF2 Modeling of Protein Complexes
Objective: Generate high-confidence structural models of proteins and their complexes for aggregation analysis.
Procedure:
- Sequence Preparation: Obtain canonical protein sequences from UniProt database (reviewed Swiss-Prot entries recommended) [69] [70].
- Multiple Sequence Alignment: Use MMseqs2 algorithm (as implemented in ColabFold) for rapid MSA construction [67].
- Structure Prediction:
- Model Selection: Rank models by predicted confidence metrics (pLDDT and predicted template modeling score [ipTM] for complexes)
- Quality Assessment: Evaluate model quality using pLDDT thresholds:
- >90: High confidence
- 70-90: Confident
- 50-70: Low confidence
- <50: Very low confidence [68]
Technical Notes: For proteins with intrinsic disorder, AF2 may produce low-confidence predictions; these regions often correspond to aggregation-prone sequences and require complementary experimental validation [71].
Experimental Protocol 2: Aggregation Risk Assessment
Objective: Identify and characterize aggregation-prone regions within protein structures.
Procedure:
- Sequence-Based Screening:
- Structure-Based Analysis:
- Map low pLDDT regions in AF2 models as potential aggregation hotspots [71]
- Calculate solvent accessibility of β-strand segments
- Identify surface-exposed hydrophobic patches
- Variant Impact Assessment:
- Cross-Seeding Potential:
- Analyze structural complementarity between different amyloidogenic proteins
- Identify potential "strain" specificities in aggregation propagation [40]
Experimental Protocol 3: Functional Validation of Aggregation-Prone Regions
Objective: Experimentally validate computational predictions of aggregation risk.
Procedure:
- In Vitro Aggregation Assays:
- Express and purify protein fragments corresponding to predicted aggregation-prone regions
- Perform Thioflavin T fluorescence kinetics to monitor fibril formation
- Analyze oligomer formation using native PAGE or size-exclusion chromatography
- Cell-Based Validation:
- Express wild-type and variant proteins in cell culture models
- Monitor protein localization and aggregate formation using immunofluorescence
- Assess cytotoxicity via MTT or LDH release assays
- Biophysical Characterization:
- Determine structural changes using circular dichroism spectroscopy
- Analyze aggregate morphology via electron microscopy
- Quantify oligomer populations using atomic force microscopy [66]
Applications in Neurodegenerative Disease Research
Case Study: Short Linear Motifs and Variant Pathogenicity
Recent research demonstrates the powerful integration of AF2 with clinical data to improve assessment of Short Linear Motifs (SLiMs) and their variant pathogenicity. The MotSASi method leverages AF2-generated structures to analyze the effects of single amino acid substitutions within SLiMs, integrating this structural analysis with population genetics data from gnomAD and clinical variant data from ClinVar [69] [70].
Table 1: Performance of AF2-Enhanced MotSASi in SLiM Prediction
| Metric | Traditional Methods | AF2-Enhanced MotSASi |
|---|---|---|
| Proteome Coverage | 10-20% (limited to PDB structures) | ~100% (all human SLiMs) |
| False Positive Rate | >80% | Significantly reduced |
| Variant Pathogenicity Prediction | Moderate accuracy | Superior to AlphaMissense |
| Applicable Motif Classes | 24 with PDB structures | 51 (24+27 without PDB structures) |
This approach demonstrates that AF2-derived structures reliably reproduce known crystallographic structures and reflect the deleterious effects of known sequence variants, enabling a comprehensive catalog of variants within SLiMs along with their deleteriousness assessments [69].
Case Study: α-Synuclein in Parkinson's Disease
In Parkinson's disease research, AF2 has been instrumental in identifying the β-strand segments (β1 and β2) of α-synuclein involved in interactions within amyloid fibrils [67]. When combined with all-atom molecular dynamics simulations, this structural information provides insights into the early oligomerization events that initiate the aggregation process.
The integration strategy involves:
- Predicting the structure of α-synuclein monomers and oligomers using AF2
- Identifying interface residues critical for aggregate propagation
- Modeling the impact of pathogenic mutations (e.g., A53T, A30P) on aggregation propensity
- Validating predictions experimentally using in vitro aggregation assays
Emerging Applications: Cross-Seeding and Strain Heterogeneity
The structural insights from AF2 are advancing understanding of cross-seeding phenomena, where aggregates of one protein can catalyze the aggregation of different pathological proteins [40]. This process may explain the frequent co-occurrence of different proteinopathies and disease heterogeneity.
Table 2: AF2 Applications in Neurodegenerative Protein Aggregation
| Disease | Primary Aggregating Protein | AF2 Application |
|---|---|---|
| Alzheimer's Disease | Aβ and tau | Modeling fibril structures, strain heterogeneity |
| Parkinson's Disease | α-synuclein | Identifying β-strand interaction interfaces |
| ALS/FTLD | TDP-43 | Predicting aggregation-prone regions in low-complexity domains |
| Alexander Disease | GFAP | Modeling Rosenthal fiber formation mechanisms |
Table 3: Essential Research Reagents and Computational Tools
| Resource | Type | Function | Application in Aggregation Research |
|---|---|---|---|
| AlphaFold2/ColabFold | Computational Tool | Protein structure prediction | Generating structural models for aggregation-prone proteins |
| ELM Database | Database | Short Linear Motif repository | Identifying potential interaction and aggregation motifs |
| FoldX | Computational Tool | Protein stability calculation | Predicting impact of variants on stability and aggregation |
| gnomAD | Database | Population genetic variation | Assessing variant frequency and constraint |
| ClinVar | Database | Clinical variant interpretations | Correlating structural predictions with pathogenicity |
| MotSASi | Computational Method | Variant deleteriousness assessment | Prioritizing pathogenic variants in SLiMs |
| Thioflavin T | Chemical Reagent | Amyloid detection | Experimental validation of fibril formation |
| Cryo-EM | Experimental Method | High-resolution structure determination | Validating AF2 predictions of aggregate structures |
Current Limitations and Future Directions
Despite its transformative impact, AF2 has several limitations in the context of aggregation risk assessment. The algorithm struggles to predict high-confidence structures for intrinsically disordered regions, which are frequently involved in protein aggregation [71]. Additionally, AF2 currently cannot reliably predict the effect of mutations on protein structure without explicitly modeling the variant sequence, and has limited capability in modeling large aggregate assemblies or the conformational dynamics of the aggregation process [71] [67].
Future developments will likely focus on:
- Improved disorder handling through integration with molecular dynamics
- Enhanced multi-chain modeling for complex aggregate structures
- Time-resolved predictions of aggregation pathways
- Integration with experimental data from cryo-EM and NMR
- Small molecule docking to identify aggregation inhibitors
The rapid pace of development in AI-based structure prediction suggests these limitations will be progressively addressed, further enhancing the utility of AF2 in neurodegenerative disease research.
The integration of AlphaFold2 with aggregation risk assessment methodologies represents a powerful paradigm shift in neurodegenerative disease research. By providing high-accuracy structural models for the entire human proteome, AF2 enables researchers to move beyond sequence-based predictions to structure-informed assessments of aggregation propensity, variant impact, and intermolecular interactions. The protocols and applications detailed in this technical guide provide a framework for leveraging these tools to advance our understanding of protein misfolding diseases and accelerate the development of targeted therapeutic strategies. As both computational and experimental methods continue to evolve, this integrated approach promises to yield increasingly insights into the structural basis of neurodegeneration and novel avenues for therapeutic intervention.
Overcoming Development Hurdles: Troubleshooting Aggregation in Biologics and Therapeutics
Addressing High-Concentration Formulation Challenges for Subcutaneous Delivery
The investigation of protein misfolding and aggregation represents a cornerstone of modern neurodegenerative disease research, providing critical insights into the pathologies of conditions such as Alzheimer's disease and Parkinson's disease. This same scientific foundation directly informs the challenges faced in developing high-concentration protein formulations for subcutaneous delivery. The same biophysical principles that govern the pathological aggregation of amyloid-β and α-synuclein in the brain—including hydrophobic interactions, electrostatic forces, and colloidal stability—also dictate the stability and viscosity of therapeutic biologic formulations [72] [73]. Understanding these shared mechanisms is essential for overcoming the delivery challenges of high-concentration monoclonal antibodies and other protein therapeutics, ultimately enabling patient-friendly administration of treatments for chronic conditions, including those affecting the central nervous system.
The pharmaceutical industry is undergoing a significant shift from intravenous to subcutaneous (SC) administration of biologics, driven by patient preference, healthcare economics, and the need for scalable treatment options in an era of rapidly expanding biologic pipelines [74]. This transition is particularly relevant for chronic neurodegenerative disorders, where frequent clinic visits for intravenous infusions present substantial burdens on patients and healthcare systems. However, developing high-concentration formulations for subcutaneous delivery presents unique technical challenges that must be addressed through advanced formulation science, analytical characterization, and innovative delivery technologies [75].
Key Challenges in High-Concentration Protein Formulations
Viscosity and Injectability
As protein concentration increases, viscosity rises exponentially, creating significant challenges for manufacturability and patient administration. High viscosity can compromise ultrafiltration/diafiltration (UF/DF) processes during manufacturing, extend production times, and potentially exceed system limitations [76]. From a patient perspective, high viscosity impacts syringeability and injectability, potentially requiring excessive injection forces that can make self-administration difficult or impossible. The molecular basis for high viscosity in concentrated antibody solutions involves a complex interplay of electrostatic and hydrophobic interactions between protein molecules [77]. These protein-protein interactions (PPI) are influenced by formulation conditions including ionic strength, pH, and excipient composition, and can lead to undesirable phenomena such as phase separation, precipitation, or gel formation [76].
Stability and Aggregation
Protein aggregation at high concentrations poses significant risks to drug product safety and efficacy. The same principles of protein misfolding that underlie neurodegenerative diseases manifest in formulation challenges, where proteins may undergo partial unfolding, surface adsorption, or colloidal instability [72] [76]. At high concentrations, the frequency of protein-protein collisions increases dramatically, raising the probability of aggregation through hydrophobic patches or specific chemical interactions. These aggregates can potentially enhance immunogenicity, reduce therapeutic efficacy, and compromise product safety [78]. Maintaining conformational stability while minimizing aggregation throughout the product shelf-life represents a critical challenge in high-concentration formulation development.
Analytical Characterization Challenges
Traditional analytical methods often require sample dilution, which can alter protein structure or aggregation behavior, providing misleading information about the true state of the high-concentration protein [76]. Accurate characterization requires orthogonal methods capable of analyzing viscous samples in their undiluted state to measure critical parameters including viscosity, turbidity, thermal stability, and syringeability. Developing stability-indicating methods that can detect subtle changes in protein conformation and interaction under high-concentration conditions is essential for ensuring consistent product quality [75].
Table 1: Key Challenges in High-Concentration Formulation Development
| Challenge Category | Specific Technical Hurdles | Impact on Development |
|---|---|---|
| Viscosity | Exponential viscosity increase with concentration; Strong protein-protein interactions | Compromised manufacturability; Limited syringeability and injectability; Potential for excessive injection force |
| Stability | Increased aggregation potential; Colloidal instability; Surface-induced denaturation | Risk of immunogenicity; Reduced therapeutic efficacy; Shorter shelf-life |
| Analytical Characterization | Dilution artifacts in traditional assays; Limited methods for undiluted analysis; Viscosity interference with measurements | Incomplete product understanding; Difficulty establishing predictive stability models |
| Delivery Device Compatibility | Limitations in injection volume (<1.5-2.0 mL for SC); Viscosity constraints for autoinjectors; Patient tolerability for large volumes | Restricted dosing options; Need for multiple injections; Complex device design requirements |
Advanced Formulation Strategies and Excipient Solutions
Viscosity Reduction Technologies
Multiple advanced strategies have been developed to address the viscosity challenges associated with high-concentration protein formulations. Approved and emerging viscosity-reducing agents (VRAs) include amino acids (particularly arginine and its derivatives), salts, and non-ionic surfactants [77]. These excipients function through various mechanisms, including modulation of electrostatic interactions, disruption of transient protein clusters, and shielding of hydrophobic patches on protein surfaces. The effectiveness of specific VRAs is highly protein-dependent, requiring systematic screening to identify optimal combinations for each molecule. Emerging approaches include synergistic combinations of excipients that target multiple interaction pathways simultaneously, potentially enabling substantial viscosity reduction without compromising stability [77].
Stabilization Approaches
Stabilization of high-concentration formulations requires a multi-pronged approach addressing both conformational and colloidal stability. Adjusting pH to maximize protein solubility while maintaining conformational integrity is a powerful strategy for reducing aggregation [76]. Optimal pH selection positions the protein sufficiently distant from its isoelectric point to enhance solubility through net charge repulsion, while avoiding extremes that might accelerate chemical degradation pathways. Sugar-based stabilizers such as sucrose and trehalose function preferentially by excluded volume effects and water replacement mechanisms, while surfactants including polysorbate 20 and 80 minimize aggregation by competing with proteins for interfaces and preventing surface-induced denaturation [78] [79]. The delicate balance between stabilizing excipients and their potential to introduce new challenges (such as surfactant-induced particle formation) requires careful optimization.
Innovative Formulation Platforms
Beyond conventional solution formulations, several advanced drug delivery systems offer alternative pathways for subcutaneous delivery of high-dose biologics. These include non-aqueous powder suspensions, which can dramatically increase the deliverable dose while maintaining low viscosity during administration [77]. In-situ forming systems, including hydrogels and precipitating formulations, provide opportunities for sustained release, potentially extending dosing intervals for chronic conditions [78]. Additionally, co-formulation with hyaluronidase enables delivery of larger volumes (typically up to 10-15 mL) by temporarily degrading subcutaneous hyaluronic acid, thereby increasing tissue permeability and dispersion [77]. Each platform presents unique advantages and development considerations that must be evaluated against the target product profile.
Table 2: Advanced Formulation Technologies for High-Concentration Subcutaneous Delivery
| Technology Platform | Mechanism of Action | Key Advantages | Development Considerations |
|---|---|---|---|
| High-Concentration Liquid Formulations | Maximizes protein concentration in solution; Optimized excipients control viscosity and stability | Direct development path; Established regulatory precedent; Compatibility with standard delivery systems | Viscosity limitations; Concentration-dependent stability challenges; May require specialized manufacturing |
| Non-Aqueous Powder Suspensions | Protein stabilization in solid state; Reconstitution at point-of-use | Ultra-high concentration potential; Improved long-term stability; Lower viscosity upon reconstitution | Additional reconstitution step; Potential bioavailability concerns; Sterility challenges for suspension formulations |
| In-Situ Forming Systems | Formation of depot after injection; Controlled release over extended periods | Reduced dosing frequency; Sustained therapeutic levels; Potential for dose sparing | Complex release kinetics; Potential for injection site reactions; More challenging bioavailability prediction |
| Hyaluronidase Co-Formulations | Temporary degradation of subcutaneous matrix; Increased tissue dispersion and volume capacity | Enables larger volume injections (up to 10-15 mL); Rapid dispersion from injection site | Enzyme compatibility with API; Potential for immunogenicity; Additional regulatory requirements for combination |
Experimental Approaches and Characterization Methods
High-Throughput Screening and Predictive Modeling
Modern formulation development leverages high-throughput screening technologies to efficiently explore multi-dimensional formulation spaces. These systems enable rapid assessment of hundreds of formulation conditions with minimal material consumption, evaluating critical quality attributes including viscosity, conformational stability, colloidal stability, and aggregation propensity [77]. Complementary to experimental approaches, in silico tools employ machine learning and molecular dynamics simulations to evaluate high-concentration suitability and formulation sensitivity early in development [76]. Starting from protein sequence alone, these computational approaches can predict susceptibility to common degradation routes, model protein-protein interactions, and screen formulation conditions (e.g., pH and excipients) to identify stabilizing environments—supporting faster, more informed formulation design and mitigating risks before committing to extensive experimental work [76].
Analytical Techniques for High-Concentration Characterization
A science-driven analytical setup employing orthogonal, stability-indicating methods is essential for comprehensive characterization of high-concentration formulations. Key techniques must be adapted to analyze samples without dilution to avoid altering protein behavior:
- Rheology: Measures viscosity and viscoelastic properties under shear rates relevant to manufacturing and injection.
- Dynamic Light Scattering (DLS): Assesses diffusion interaction parameter (kD) and colloidal stability in concentrated solutions.
- Differential Scanning Calorimetry (DSC): Evaluates conformational stability through thermal unfolding transitions.
- UV-Vis and Static Light Scattering: Determines turbidity and opalescence as indicators of phase behavior.
- Size Exclusion Chromatography (SEC-UHPLC): Quantifies soluble aggregates after appropriate dilution, with careful attention to potential dilution artifacts.
- Microflow Imaging (MFI) and Light Obscuration: Characterizes subvisible and visible particles.
These methods collectively provide a comprehensive understanding of protein behavior under high-concentration conditions, enabling evidence-based formulation decisions and robust control strategy development.
Protocol: Comprehensive Formulation Screening for Viscosity and Stability Optimization
Objective: Systematically evaluate formulation parameters to identify optimal conditions for high-concentration protein formulations balancing viscosity, stability, and manufacturability requirements.
Materials and Equipment:
- Purified protein drug substance (>95% purity)
- Excipient screening library (buffers, salts, amino acids, surfactants, sugars)
- High-throughput formulation platform (e.g., Tecan or Hamilton liquid handling system)
- Rheometer (cone-plate or capillary viscometer)
- Dynamic light scattering instrument
- Differential scanning calorimeter
- Stability chambers for accelerated stability studies
Procedure:
Design of Experiments (DoE) Setup
- Define critical formulation factors: pH (5.0-7.0), ionic strength (0-150 mM), primary stabilizer (sucrose, trehalose, 0-10% w/v), and surfactant concentration (0-0.1% w/v)
- Create response surface model with 40-50 formulation permutations
High-Throughput Formulation Preparation
- Use liquid handling system to prepare 1-2 mL of each formulation condition
- Concentrate to target protein concentration (100-150 mg/mL) using centrifugal concentrators
- Filter sterilize (0.22 μm) and aliquot for various characterization assays
Viscosity Assessment
- Measure viscosity at shear rates from 100-10,000 s⁻¹ using cone-plate rheometer at 25°C
- Record apparent viscosity at 10,000 s⁻¹ (representative of injection shear rate)
- Classify formulations as acceptable (<20 cP), borderline (20-50 cP), or unacceptable (>50 cP)
Stability Evaluation
- Conduct short-term (2-4 weeks) accelerated stability at 25°C/60% RH and 40°C/75% RH
- Monitor visual appearance, subvisible particles, soluble aggregates (SEC), and fragmentation (CE-SDS)
- Assess conformational stability by DSC (Tm1, Tm2, Tagg)
Data Analysis and Selection
- Construct response surface models for each critical quality attribute
- Identify design space meeting all target attributes
- Select 3-5 lead formulations for further characterization
Expected Outcomes: Identification of formulation conditions providing optimal balance of low viscosity (<20 cP) and adequate stability (<2% aggregate formation after 4 weeks at 40°C) for further development.
The Scientist's Toolkit: Essential Research Reagents and Technologies
Table 3: Research Reagent Solutions for High-Concentration Formulation Development
| Reagent Category | Specific Examples | Function & Application | Technical Considerations |
|---|---|---|---|
| Viscosity-Reducing Agents | L-arginine HCl, Sodium chloride, Glycine | Modulate protein-protein interactions; Reduce viscosity through electrostatic shielding | Concentration-dependent effects; Potential impact on conformational stability; May influence opalescence |
| Stabilizing Excipients | Sucrose, Trehalose, Sorbitol | Preferentially exclude proteins from solution; Stabilize native conformation | Concentration typically 5-10% (w/v); Potential for high viscosity at concentrations >10% |
| Surfactants | Polysorbate 20, Polysorbate 80 | Minimize surface-induced aggregation; Stabilize against interfacial stress | Quality and grade critical for biopharma; Peroxide formation in polysorbates requires monitoring |
| Buffers | Histidine, Succinate, Citrate, Phosphate | Control formulation pH; Provide chemical stability | Buffer capacity and concentration optimization; Potential for crystallization at low temperatures |
| Novel Excipients | Hydroxypropyl-β-cyclodextrin, Sulfobutylether-β-cyclodextrin | Form inclusion complexes; Enhance solubility of hydrophobic proteins | Require extensive safety profiling; Regulatory considerations for novel excipients |
Device Integration and Patient Considerations
Delivery Device Technologies
Successful subcutaneous delivery of high-concentration biologics requires careful integration of drug product with appropriate delivery device technology. Multiple device options exist along a spectrum of complexity and patient interaction:
Prefilled Syringes with UTW Needles: Conventional prefilled syringes equipped with shorter ultra-thin wall (UTW) or tapered needles can reduce injection pain and accommodate moderate viscosities (<50 cP) [77]. These systems represent the simplest approach for patient self-administration.
Autoinjectors: Handheld autoinjectors provide automated delivery, simplifying administration for patients with dexterity or vision challenges. Current generation devices can typically handle volumes up to 2.0 mL and viscosities below 50 cP, though next-generation devices targeting higher viscosity capabilities are in development [75].
Wearable Devices: On-body delivery systems (OBDS) or patch pumps enable delivery of larger volumes (typically 2-10 mL) over extended periods (minutes to hours), distributing the volume over time to enhance tolerability [77]. These systems are particularly valuable for high-dose therapies exceeding conventional volume limitations.
Device selection requires careful consideration of drug product characteristics (viscosity, volume), patient capabilities, and commercial requirements, with human factors studies playing a critical role in identifying optimal user interfaces [75].
Patient-Centric Development
The ultimate success of any high-concentration subcutaneous product depends on patient acceptance and adherence. Development programs must incorporate patient perspectives through well-designed preference studies and human factors testing [75]. Key considerations include injection volume tolerability (typically <1.5 mL preferred, up to 2.0 mL acceptable), injection time (<10-20 seconds for bolus injections), overall treatment burden, and device usability [74]. For chronic conditions, such as neurodegenerative disorders, these factors significantly influence long-term adherence and treatment outcomes. The definition of quality target product profile (QTPP) should explicitly incorporate patient preferences regarding dosing frequency, administration time, and device complexity to ensure development of truly patient-centric therapies [75].
The development of high-concentration formulations for subcutaneous delivery represents a critical enabler for the next generation of biologic therapies, particularly relevant for chronic conditions requiring long-term treatment. By addressing the fundamental challenges of viscosity, stability, and delivery through integrated formulation, device, and manufacturing strategies, developers can create transformative treatments that reduce healthcare system burdens while improving patient quality of life. The continued advancement of in silico prediction tools, high-throughput screening methodologies, and innovative formulation platforms will accelerate this paradigm shift from intravenous to patient-friendly subcutaneous administration, ultimately making advanced therapies more accessible across diverse patient populations and healthcare settings.
The development of advanced biologic therapeutics, including antibody-drug conjugates (ADCs) and viral vectors, represents a transformative approach in modern medicine, particularly for treating cancer and genetic disorders. These complex modalities face significant stability challenges during formulation development and storage, primarily due to their susceptibility to physical degradation and chemical instability. Within the broader context of protein misfolding and aggregation research—a central theme in neurodegenerative diseases such as Alzheimer's and Parkinson's—the stability issues of biotherapeutics reveal striking parallels in their fundamental mechanisms. Protein aggregation, characterized by the misfolding and self-assembly of proteins into toxic oligomers and fibrils, is not only a hallmark of neurological pathology but also a critical barrier to the development of stable, efficacious biologics [16] [11] [80].
The stability profiles of ADCs and viral vectors are governed by similar principles of protein homeostasis that are compromised in neurodegenerative conditions. Conformational instability (alterations in protein structure) and colloidal instability (tendency to aggregate) adversely affect product quality, shelf life, and therapeutic efficacy [81]. For viral vectors, the brittleness of viral capsids and sensitivity to environmental stresses mirror the fragility of native protein folding states, while ADCs face additional complexities from conjugation chemistry that can destabilize the antibody structure [81] [82]. Understanding these challenges through the lens of protein misfolding provides valuable insights for developing stabilization strategies that maintain the intrinsic properties of these advanced therapeutics from manufacturing through clinical administration.
Stability Challenges in Antibody-Drug Conjugates (ADCs)
Key Stability Concerns and Underlying Mechanisms
ADCs represent a sophisticated class of biotherapeutics designed to selectively deliver potent cytotoxic agents to target cells. However, their structural complexity, comprising a monoclonal antibody, chemical linker, and highly active payload, introduces multiple potential failure points that can compromise stability and efficacy.
Linker Instability: The chemical bridge connecting the antibody to the payload is critically susceptible to cleavage, potentially leading to premature release of the cytotoxic drug before reaching the target tissue. This not only reduces therapeutic efficacy but also increases systemic toxicity [81] [82]. Different linker classes exhibit distinct vulnerability profiles: acid-labile linkers (e.g., those in gemtuzumab ozogamicin) may hydrolyze in acidic environments; protease-cleavable linkers are susceptible to enzymatic degradation; and disulfide-based linkers can be affected by redox conditions [82].
Aggregation Propensity: The conjugation of hydrophobic payloads to antibodies significantly increases molecular hydrophobicity, driving colloidal instability and aggregation [81] [83]. Organic solvents used during conjugation reactions further exacerbate this tendency. These aggregates can impact product quality, reduce potency, and increase immunogenicity risk, presenting significant challenges for manufacturing and storage [83].
Conformational Destabilization: The attachment of payload molecules can alter the structural integrity of the monoclonal antibody, potentially leading to partial unfolding, increased susceptibility to proteolytic degradation, and exposure of hydrophobic regions that normally remain buried within the native structure [81]. This destabilization shares mechanistic similarities with the initial protein misfolding events observed in neurodegenerative pathologies [11] [80].
Drug-Antibody Ratio (DAR) Heterogeneity: Stochastic conjugation methods typically produce heterogeneous mixtures with varying numbers of drug molecules per antibody. Species with exceptionally high DAR values often demonstrate reduced stability and accelerated clearance, while low-DAR variants exhibit diminished potency [82] [84]. This heterogeneity complicates pharmacokinetic profiling and dose optimization.
Table 1: Key Stability Challenges in Antibody-Drug Conjugates
| Challenge | Impact on Product Quality | Root Cause |
|---|---|---|
| Linker Instability | Premature payload release; Reduced efficacy; Increased toxicity | Susceptibility to pH, enzymatic cleavage, or redox conditions |
| Aggregation | Increased immunogenicity; Reduced activity; Product loss | Enhanced hydrophobicity from payload; Conjugation process |
| Conformational Instability | Altered pharmacokinetics; Potential loss of target binding | Structural perturbation from conjugation |
| DAR Heterogeneity | Inconsistent potency; Variable pharmacokinetics | Non-specific conjugation chemistry |
Experimental Protocols for ADC Stability Assessment
Protocol for Aggregate Removal Using Membrane Chromatography
The following methodology details the optimization of aggregate removal from ADCs using anion-exchange membrane chromatography, based on design-of-experiments (DoE) principles [83].
Materials and Reagents:
- Sartobind Q Nano capsules (1 mL and 3 mL membrane volumes)
- ADC material produced from stochastic conjugation on IgG1
- Phosphate buffer (20 mM), pH range 7.4-8.6
- Sodium chloride (1M) for regeneration
- Sodium hydroxide (1M) for sanitization
- Dimethylacetamide (DMAC) for solvent conditioning
Equipment:
- NanoDrop spectrophotometer (Thermo Fisher Scientific) for concentration measurement
- HPLC system with size exclusion column for aggregate quantification
- High-resolution mass spectrometer for DAR determination
- Conductivity and pH meters
Procedure:
- Membrane Preparation: Sanitize the membrane capsule with 1M NaOH solution, followed by regeneration with 1M NaCl. Equilibrate with 20 mM phosphate buffer at the desired pH (7.4-8.6) and conductivity (1-5 mS/cm).
- Sample Loading: Load the ADC solution (1-10 g/L concentration) onto the membrane at flow rates of 5-10 membrane volumes per minute. Maintain the load between 20-60 g/L sorbent.
- Product Collection: Collect flow-through fractions and analyze for ADC concentration, HMW content, and DAR distribution.
- Process Optimization: Utilize a D-optimal design with factors including pH (8.1-8.5), conductivity (1-4 mS/cm), and ADC concentration (1-10 g/L). Response variables should include HMW clearance, yield, and DAR maintenance.
- Analytical Methods:
- Quantify ADC concentration by UV spectroscopy at 280 nm
- Determine HMW species by SEC-HPLC using a TSKgel G3000SWXL column
- Calculate DAR by UV spectroscopy, SEC-HPLC, and HRMS
Key Findings: Optimal conditions for aggregate clearance were achieved at pH >8.4 with conductivity >3.4 mS/cm, providing >80% HMW removal while maintaining yields >90% and target DAR [83].
Protocol for Assessing Linker Stability
Materials and Reagents:
- ADC reference standard
- Plasma or serum (human or species-specific)
- Buffer solutions at varying pH (4.0-7.4)
- Proteolytic enzymes (e.g., cathepsin B for protease-cleavable linkers)
- Acetonitrile and formic acid for HPLC analysis
Equipment:
- HPLC system with reverse-phase column
- Mass spectrometer for payload identification
- Incubators maintained at 37°C
Procedure:
- Plasma Stability Study: Incubate ADC in plasma at 37°C. Withdraw samples at predetermined time points (0, 6, 24, 48, 72 hours). Precipitate proteins with acetonitrile and analyze supernatant for free payload using LC-MS/MS.
- pH Stability Assessment: Prepare buffers at physiologically relevant pH values (4.0 for endosomal pH, 7.4 for physiological pH). Incubate ADC samples at 37°C and analyze for linker cleavage and payload release over time.
- Enzymatic Stability: For protease-cleavable linkers, incubate ADC with relevant enzymes (e.g., cathepsin B) and monitor cleavage products.
- Data Analysis: Calculate half-life of ADC and percentage of payload released under each condition to determine linker stability profile.
ADC Stability Assessment Workflow
Stability Challenges in Viral Vectors
Key Stability Concerns by Vector Type
Viral vectors constitute essential delivery platforms for gene therapy applications, but they present diverse and significant stability challenges that vary by vector class. Maintaining the functional integrity of these complex biologics from production through clinical administration requires careful consideration of their distinct vulnerability profiles.
Adeno-Associated Virus (AAV) Vectors: While AAVs offer favorable safety profiles and sustained gene expression capabilities, their capsid integrity is susceptible to pH fluctuations, temperature variations, and freeze-thaw stress [81] [85]. The single-stranded DNA genome within AAV particles can undergo chemical degradation, including depurination and strand breakage, while the capsid itself may suffer from conformational changes that reduce cellular transduction efficiency.
Adenoviral Vectors: These vectors exhibit sensitivity to both thermal and mechanical stresses. Their propensity to trigger immune responses presents an additional stability challenge, as pre-existing immunity can neutralize the vector before it reaches target cells [81] [85]. First-generation adenoviral vectors accommodate transgenes up to 6.5 kb but elicit significant immunological reactions, while later generations with reduced viral gene expression offer improved stability profiles.
Lentiviral and Retroviral Vectors: As enveloped viruses, lentiviral and retroviral vectors demonstrate particular vulnerability to shear stress, freeze-thaw cycles, and temperature fluctuations. The lipid bilayer envelope can undergo phase transitions or fusion events that compromise vector functionality [81] [85]. Additionally, gamma-retroviral vectors have been associated with insertional mutagenesis due to random genomic integration, a stability concern at the genetic level.
Table 2: Stability Profiles of Major Viral Vector Platforms
| Vector Type | Primary Stability Challenges | Impact on Function |
|---|---|---|
| Adeno-Associated Virus (AAV) | Capsid instability; DNA degradation; Sensitivity to pH and temperature | Reduced transduction efficiency; Loss of genome integrity |
| Adenovirus | Immunogenicity; Thermal instability; Mechanical stress sensitivity | Vector neutralization; Loss of infectivity |
| Lentivirus/Retrovirus | Envelope fragility; Shear sensitivity; Freeze-thaw instability | Decreased titer; Loss of transduction capacity |
| Herpes Simplex Virus | Large genome susceptibility; Complex structure instability | Challenges in manufacturing consistency |
Experimental Protocols for Viral Vector Stability Assessment
Protocol for Vector Titer and Potency Stability Studies
Materials and Reagents:
- Viral vector sample (AAV, adenovirus, lentivirus, etc.)
- Permissive cell line for vector propagation (e.g., HEK293 for AAV)
- Culture media and supplements
- DNA extraction kit
- qPCR reagents including primers for vector genome quantification
- Transduction reagents (polybrene for retrovirus/lentivirus)
Equipment:
- qPCR instrument
- Cell culture facility with CO2 incubators
- Flow cytometer (for functional assays)
- Thermal cycler
Procedure:
- Physical Titer Determination:
- Extract vector genome using DNA extraction kit
- Perform qPCR with primers specific to vector sequence (e.g., polyA region)
- Compare to standard curve of known concentration to determine vector genome (vg/mL)
- Store aliquots at different conditions (-80°C, -20°C, 4°C, room temperature) and test at timepoints (0, 1, 3, 6 months)
Functional Potency Assay:
- Seed permissive cells in multi-well plates
- Transduce with serial dilutions of vector sample
- Incubate for appropriate duration (varies by vector system)
- For reporter vectors: analyze expression by flow cytometry or fluorescence microscopy
- For therapeutic vectors: measure relevant functional output (e.g., protein expression by ELISA)
- Calculate functional titer (transducing units/mL)
Capsid Integrity Assessment (for AAV):
- Perform ELISA with AAV capsid antibodies
- Use electron microscopy to visualize particle morphology
- Conduct charge detection mass spectrometry for empty/full capsid ratio
Data Analysis: Plot retention of physical and functional titers over time under various storage conditions. Calculate degradation rates and identify critical stability parameters.
Protocol for Excipient Screening to Enhance Vector Stability
Materials and Reagents:
- Viral vector preparation
- Candidate stabilizers (surfactants, sugars, polymers, antioxidants)
- Formulation buffers
- Cryoprotectants (for freeze-thaw studies)
Equipment:
- Thermal cycler for controlled freeze-thaw
- Dynamic light scattering instrument for particle size
- Differential scanning calorimeter
Procedure:
- Formulation Preparation: Dialyze or dilute vector into candidate formulation buffers containing various excipients (e.g., sucrose, trehalose, poloxamers, histidine buffers).
- Stress Testing:
- Thermal Stress: Incubate formulations at 4°C, 25°C, and 37°C for predetermined periods
- Freeze-Thaw Cycling: Subject samples to repeated freeze-thaw cycles (-80°C to room temperature)
- Mechanical Stress: Agitate samples using orbital shaker or subject to shear stress
- Analysis:
- Measure recovery of functional titer after stress
- Assess particle aggregation by dynamic light scattering
- Evaluate capsid integrity by ELISA or electron microscopy
- DoE Optimization: Use statistical experimental design to optimize excipient combinations and concentrations based on multiple stability responses.
Viral Vector Stability Assessment Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Stability Optimization Studies
| Reagent/Material | Function in Stability Research | Example Applications |
|---|---|---|
| Sartobind Q Membrane | Anion-exchange chromatography for aggregate removal | ADC polishing step; HMW species clearance [83] |
| Size Exclusion HPLC Columns | Separation and quantification of aggregates and fragments | Analysis of oligomeric states in ADCs and viral vectors [83] |
| Dynamic Light Scattering Instrument | Hydrodynamic size measurement and aggregation assessment | Viral vector particle integrity; ADC self-association [85] |
| qPCR System | Vector genome quantification and integrity assessment | Viral vector titer determination; Genome stability studies [85] |
| Plasma/Serum Samples | Biological matrix for stability assessment | Linker stability in physiological conditions [82] |
| Stabilizing Excipients | Prevention of aggregation and degradation | Lyophilization protectants; Storage buffer additives [81] |
| LC-MS/MS System | Payload identification and quantification | Linker cleavage analysis; Metabolite identification [82] [84] |
Strategic Stabilization Approaches and the Protein Quality Control Connection
The stabilization of advanced biotherapeutics shares fundamental principles with cellular protein quality control mechanisms that prevent aggregation in neurodegenerative diseases. By understanding these parallels, researchers can develop more effective stabilization strategies for both ADCs and viral vectors.
Molecular Chaperones and Stabilization Excipients
Cells employ molecular chaperones to prevent protein misfolding and aggregation, a system that inspires therapeutic stabilization approaches. For ADCs, excipients such as sucrose, trehalose, and histidine act as chemical chaperones by stabilizing native protein conformation through preferential exclusion mechanisms [81]. These stabilizers strengthen the colloidal stability of ADCs, reducing aggregation propensity during storage and administration. Similarly, viral vectors benefit from excipients that mimic chaperone functions—sugars and polyols stabilize capsid proteins, while surfactants (e.g., poloxamers) prevent air-liquid interface-induced denaturation [81] [85]. The concentration and combination of these stabilizers can be optimized using quality by design (QbD) principles, similar to how cells regulate chaperone expression through heat shock factor 1 (HSF1) activation in response to proteotoxic stress [11] [66].
Clearance Mechanisms and Aggregate Removal Strategies
Cellular systems employ sophisticated clearance mechanisms—including the ubiquitin-proteasome system and autophagy—to degrade misfolded proteins and aggregates [11] [80]. Similarly, biomanufacturing processes implement purification strategies to remove aggregates from ADC and viral vector products. Membrane chromatography technologies function analogously to selective autophagy by specifically recognizing and removing high molecular weight species while allowing monomeric products to pass through [83]. The optimization of pH and conductivity parameters in membrane chromatography mirrors the regulation of lysosomal pH for optimal hydrolase activity in cellular clearance pathways. For viral vectors, chromatographic methods that separate empty from full capsids emulate the cellular quality control systems that distinguish properly assembled complexes from defective ones [85].
Conformational Stabilization and Linker Optimization
The stabilization of native protein structures represents another key strategy shared between neurodegenerative disease therapeutics and biologics manufacturing. In transthyretin amyloidosis, the drug tafamidis prevents misfolding by stabilizing the native tetrameric structure [65]. Similarly, strategic engineering of ADC linkers and conjugation sites enhances structural stability—site-specific conjugation methods create more homogeneous products with improved stability profiles compared to stochastic conjugation [82] [84]. For viral vectors, engineering approaches that introduce stabilizing mutations into capsid proteins parallel the natural evolutionary optimization of protein folding, resulting in vectors with enhanced thermal stability and reduced aggregation propensity [81] [85].
Stabilization Mechanisms: Biological Inspiration and Biotech Applications
The optimization of stability for advanced biotherapeutics represents a critical challenge in modern drug development, with ADCs and viral vectors posing distinct but interconnected stability concerns. Through carefully designed experimental protocols and strategic implementation of stabilization approaches inspired by natural protein quality control systems, researchers can significantly enhance the manufacturability, shelf life, and therapeutic performance of these complex modalities. The conceptual framework of protein misfolding and aggregation—fundamental to understanding neurodegenerative diseases—provides valuable insights for addressing stability challenges in biologics development. As the field advances, the integration of sophisticated analytical methods, quality by design principles, and biologically-inspired stabilization strategies will continue to drive improvements in product quality and patient outcomes across diverse therapeutic areas.
Proteostatic collapse represents a critical pathological state in which the cellular machinery governing protein homeostasis becomes overwhelmed, leading to the accumulation of misfolded and aggregated proteins. Within the context of neurodegenerative diseases, this phenomenon drives the progression of conditions such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS). This technical review examines the molecular underpinnings of proteostatic collapse, focusing on the delicate balance between protein synthesis, folding, and degradation pathways. We present quantitative analyses of proteostasis network disruptions across disease states, detailed experimental methodologies for investigating proteostasis mechanisms, and visualization of key pathways implicated in neurodegenerative proteinopathies. The synthesis of current research provides a framework for developing targeted therapeutic strategies aimed at restoring proteostatic balance in neuronal cells.
The maintenance of protein homeostasis, or proteostasis, is essential for cellular health and function, particularly in long-lived, post-mitotic neurons [10] [86]. The proteostasis network (PN) encompasses an integrated system of approximately 2,000 components in human cells that coordinate protein synthesis, folding, trafficking, and degradation [86]. In neurodegenerative diseases, the collapse of this network results in the accumulation of toxic, misfolded protein aggregates that drive neuronal dysfunction and cell death [10] [12]. A hallmark of these conditions is the presence of disease-specific protein aggregates: amyloid-β and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, and TDP-43 in amyotrophic lateral sclerosis [10] [11]. These aggregates not only disrupt neuronal function directly but also sequester essential proteostasis components, creating a self-reinforcing cycle of proteostatic collapse [10] [12].
The vulnerability of neurons to proteostatic collapse stems from their unique biological characteristics, including high metabolic activity, complex morphology, and limited regenerative capacity [87]. Aging represents the primary risk factor for most neurodegenerative diseases, coinciding with a documented decline in PN capacity across model organisms and human tissues [86]. Understanding the molecular mechanisms governing proteostasis balance and developing strategies to reinforce this network represents a promising therapeutic approach for neurodegenerative proteinopathies.
Molecular Architecture of the Proteostasis Network
Core Components and Pathways
The proteostasis network comprises three principal modular branches that function in concert to maintain proteome integrity: (1) protein synthesis and translational control, (2) folding and conformational maintenance by molecular chaperones, and (3) protein degradation via the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) [86]. These systems are highly interconnected, with molecular chaperones serving as central coordinators across all functional modules [86].
Molecular chaperones play indispensable roles in protein quality control by facilitating de novo folding, preventing aggregation, refolding misfolded proteins, and targeting irreparably damaged proteins for degradation [1]. The major chaperone families include Hsp70, Hsp90, and chaperonins (Hsp60), each with specialized functions in protein maturation and quality control [86] [1]. The discovery of molecular chaperones emerged from studies of cellular stress responses, beginning with Ritossa's 1962 observation of chromosomal "puffs" in heat-shocked fruit flies, which corresponded to induced expression of heat shock proteins (HSPs) [1].
Table 1: Major Molecular Chaperone Families in Neuronal Proteostasis
| Chaperone Family | Representative Members | Primary Functions | Role in Neurodegeneration |
|---|---|---|---|
| Hsp70 | HSC70, HSPA8 | De novo folding, refolding, CMA substrate recognition | Mutations linked to peripheral neuropathies; CMA dysfunction in PD |
| Hsp90 | HSP90α, HSP90β | Maturation of kinase and steroid receptor clients | Stabilizes pathological tau conformations in AD |
| Chaperonins | TRiC/CCT, Hsp60 | Folding of actin, tubulin, and other complex proteins | Facilitates huntingtin aggregation in polyQ diseases |
| Small HSPs | Hsp27, αB-crystallin | Prevention of aggregation, stress resistance | Protective against α-synuclein and Aβ toxicity |
| J-domain proteins | DNAJB6, DNAJB8 | Hsp70 co-chaperones, regulate ATPase activity | Suppresses polyQ aggregation; mutations in ALS |
Protein Degradation Pathways
Cellular protein degradation occurs primarily through two coordinated systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP). The UPS is responsible for the rapid degradation of short-lived and soluble proteins, while autophagy handles larger structures, including protein aggregates and damaged organelles [86]. Within autophagy, three distinct forms have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [88] [89].
CMA is particularly relevant to neurodegenerative disease due to its selectivity and role in degrading specific pathological proteins. Unlike other forms of autophagy, CMA does not involve vesicular formation but instead relies on direct translocation of substrate proteins across the lysosomal membrane via the LAMP-2A receptor [88] [89]. Approximately 30% of cytosolic proteins contain the KFERQ-like motif required for CMA degradation, including several proteins implicated in neurodegeneration [89]. CMA activity declines with age, potentially contributing to the accumulation of aggregation-prone proteins in older individuals [88].
Quantitative Analysis of Proteostasis Network Dysregulation in Disease
Large-scale pan-disease analyses reveal distinct patterns of proteostasis network disruption across neurodegenerative conditions. A comprehensive study examining 32 human diseases across 7 categories demonstrated that proteostasis proteins are significantly over-represented in disease gene sets, with particularly strong associations in cancer and neurodegenerative diseases [90]. Proteostasis proteins comprise 25-36% of disease gene sets in cancer and 30-35% in neurodegenerative diseases, underscoring their central role in pathogenesis [90].
Table 2: Proteostasis Pathway Enrichment Across Neurodegenerative Diseases
| Disease | ALP Enrichment | UPS Enrichment | Extracellular Proteostasis | Molecular Chaperones |
|---|---|---|---|---|
| Alzheimer's Disease | +++ | +++ | +++ | +++ |
| Parkinson's Disease | +++ | +++ | +++ | ++ |
| Huntington's Disease | ++ | + | + | + |
| Amyotrophic Lateral Sclerosis | ++ | ++ | + | ++ |
| Prion Diseases | +++ | + | +++ | ++ |
+++ strongly enriched, ++ moderately enriched, + weakly enriched Data adapted from Lim & Vendruscolo, 2025 [90]
Three distinct proteostasis states have been identified that differentiate disease types [90]:
- State 1: Characterized by significant UPS perturbation with limited extracellular proteostasis involvement (typical in cancers)
- State 2: Involves extensive perturbation of both UPS and extracellular proteostasis (predominant in neurodegenerative diseases)
- State 3: Features distinctive deregulation of extracellular proteostasis with limited UPS involvement (common across autoimmune, endocrine, and cardiovascular diseases)
The neurodegenerative proteostasis state (State 2) reflects the complex protein aggregation landscape in these disorders, involving both intracellular aggregation (handled primarily by UPS) and extracellular deposition (requiring extracellular quality control mechanisms) [90] [11].
Experimental Models and Methodologies for Proteostasis Research
Co-culture Systems for Neuronal Proteostasis Studies
Advanced model systems have been developed to investigate cell-type-specific responses in proteostasis dynamics. A recently established dual-species co-culture model of human excitatory neurons and mouse glia enables precise quantification of cell-type-specific, age-related changes in the proteostasis network using data-independent acquisition (DIA) LC-MS/MS proteomics [87]. This system permits tracking of branch-specific unfolded protein response (UPR) activation by monitoring curated effector proteins downstream of the ATF6, IRE1/XBP1s, and PERK pathways [87].
Experimental Protocol: Co-culture Proteostasis Analysis
- Cell Culture Establishment: Differentiate human excitatory neurons from induced pluripotent stem cells (iPSCs) and culture with primary mouse glial cells in compartmentalized systems that allow shared medium while maintaining cellular segregation.
- Aging Induction: Subject cultures to prolonged maintenance (60+ days) with periodic stress induction using sublethal proteotoxic insults (e.g., low-dose proteasome inhibitors, mild oxidative stress).
- Stimulus Application: Treat aged cultures with pharmacological ER stressors including tunicamycin (2.5 μg/mL, 8h), thapsigargin (300 nM, 12h), or Brefeldin A (5 μM, 16h).
- Protein Extraction and Preparation: Harvest cells using species-specific antibodies for separation, followed by lysis in urea buffer (8M urea, 50mM Tris-HCl pH8, 75mM NaCl) with protease and phosphatase inhibitors.
- Proteomic Analysis: Perform data-independent acquisition LC-MS/MS on a Q-Exactive HF-X mass spectrometer with a 90-minute gradient. Process data using Spectronaut (v15) against species-specific reference databases.
- Pathway Activation Quantification: Calculate UPR branch activation by monitoring effector proteins including BiP, CHOP, XBP1s, ATF4, and phospho-eIF2α.
This approach has revealed that aging neurons largely preserve proteostasis capacity through ATF6 branch activation and maintenance of XBP1s and PERK pathways, while glia exhibit broad downregulation of proteostasis factors during aging [87].
CMA Activity Assays
Multiple methodologies have been developed to quantitatively assess chaperone-mediated autophagy activity, essential for understanding its role in neurodegenerative processes.
Experimental Protocol: CMA Activity Measurement
- Lysosomal Isolation: Prepare intact lysosomes from tissue samples or cultured cells using discontinuous Percoll density gradient centrifugation.
- CMA Substrate Preparation: Isolate radiolabeled cytosolic proteins (e.g., GAPDH) or use recombinant KFERQ-containing proteins tagged with fluorophores.
- Uptake Assay: Incubate lysosomes (50-100 μg protein) with CMA substrates (5-10 μg) in 0.3M sucrose, 10mM MOPS-KOH (pH7.3), 5mM MgCl₂, 5mM DTT, and ATP-regenerating system (2mM ATP, 10mM phosphocreatine, 10μg creatine phosphokinase) for 20-40 minutes at 37°C.
- Degradation Quantification: Measure protein degradation by TCA precipitation of non-degraded proteins or fluorescence detection of cleaved tags.
- CMA Activation Assessment: Use a CMA-specific fluorescent reporter (photoactivable KFERQ-dendra) to visualize and quantify lysosomal CMA activity as fluorescent puncta formation.
This methodology has demonstrated that CMA is upregulated in response to genotoxic stress, with maximal activation reached at 12 hours post-insult and remaining elevated at 24 hours [91]. Furthermore, chemical enhancement of CMA using compounds like AR7 (a retinoic acid derivative) significantly improves cellular viability upon genotoxic challenge and reduces DNA damage load [91].
Visualization of Key Proteostasis Pathways
Chaperone-Mediated Autophagy (CMA) Pathway
Diagram 1: Chaperone-Mediated Autophagy (CMA) Mechanism. This pathway illustrates the sequential process whereby cytosolic proteins containing KFERQ motifs are selectively targeted for lysosomal degradation.
Proteostasis Network Organization
Diagram 2: Proteostasis Network Integration. This diagram illustrates the interconnected nature of protein homeostasis mechanisms and how their disruption leads to proteostatic collapse.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Proteostasis Investigation
| Reagent/Category | Specific Examples | Research Application | Experimental Function |
|---|---|---|---|
| CMA Modulators | AR7, LAMP-2A overexpression vectors | CMA pathway studies | Selective activation of chaperone-mediated autophagy |
| Lysosomal Inhibitors | Bafilomycin A1, Chloroquine | Autophagy flux assays | Inhibition of lysosomal acidification and function |
| Proteasome Inhibitors | MG132, Bortezomib | UPS function studies | Reversible and irreversible proteasome inhibition |
| ER Stress Inducers | Tunicamycin, Thapsigargin | UPR pathway analysis | Induction of endoplasmic reticulum stress |
| Molecular Chaperone Antibodies | Anti-HSC70, Anti-HSP90, Anti-LAMP-2A | Protein localization and expression | Detection of chaperone expression and compartmentalization |
| CMA Reporters | KFERQ-dendra, KFERQ-PA-GFP | CMA activity measurement | Photoactivatable substrates for CMA tracking |
| Autophagy Modulators | Rapamycin, 3-Methyladenine | Macroautophagy studies | mTOR inhibition and autophagy regulation |
| Genotoxic Agents | Etoposide, Methylmethanesulfonate | DNA damage-CMA connection studies | Induction of DNA damage to examine CMA response |
Therapeutic Strategies Targeting Proteostatic Collapse
Current therapeutic approaches for rebalancing proteostasis in neurodegenerative diseases focus on multiple nodes within the proteostasis network. These include:
1. Chaperone Modulation: Enhancing the expression or function of molecular chaperones represents a promising strategy for preventing protein misfolding and aggregation. Induction of heat shock proteins through Hsf1 activation has demonstrated protective effects in models of polyglutamine diseases and synucleinopathies [10] [1].
2. Degradation Pathway Enhancement: Both UPS and autophagy pathways offer therapeutic targets. Small molecule enhancers of proteasome activity and CMA activators like AR7 show potential for clearing aggregated proteins [91]. Notably, CMA degradation targets several neurodegenerative disease proteins, including α-synuclein and tau, making its enhancement particularly relevant [88] [89].
3. Unfolded Protein Response Modulation: The UPR represents a key signaling node connecting ER proteostasis to overall cellular health. Selective modulation of specific UPR branches (PERK, IRE1, ATF6) rather than pan-UPR inhibition may provide optimal therapeutic benefit while minimizing toxicity [87] [11].
4. Combination Approaches: Given the interconnected nature of the proteostasis network, combined approaches targeting multiple pathways simultaneously may yield superior results compared to single-target interventions. Examples include coupling Hsp90 inhibition with autophagy induction or proteasome enhancement with chaperone overexpression [11] [1].
Recent research has revealed that proteostasis pathways previously considered primarily in quality control contexts, such as CMA, play surprisingly diverse roles in cellular regulation. For instance, CMA participates in genome stability maintenance through regulated degradation of checkpoint kinase 1 (Chk1) following DNA damage [91]. This expanded understanding of proteostasis network functions underscores its fundamental importance in cellular homeostasis and the potential broad benefits of its therapeutic modulation.
Proteostatic collapse represents a central pathological mechanism in neurodegenerative diseases, driven by an imbalance between protein production, folding capacity, and degradation systems. The intricate connections between components of the proteostasis network create both challenges and opportunities for therapeutic intervention. Quantitative analyses reveal distinct proteostasis signatures across neurodegenerative conditions, suggesting both common and disease-specific pathogenic mechanisms. Advanced experimental models, including neuronal-glial co-culture systems and precise activity assays for pathways like CMA, provide increasingly sophisticated tools for dissecting these complex processes. Therapeutic strategies that target multiple nodes within the proteostasis network, particularly those enhancing protein degradation capacity and bolstering chaperone function, hold significant promise for addressing the fundamental protein folding defects that drive neurodegeneration. As our understanding of proteostasis mechanisms continues to expand, so too will opportunities for developing effective interventions for these devastating disorders.
Mitigating Seeding and Cross-Seeding Between Different Pathological Proteins
The propagation of pathology in neurodegenerative diseases is fundamentally driven by the prion-like behavior of several key pathogenic proteins. This process, known as seeding, involves the template-directed misfolding of native proteins into pathological aggregates by a pre-existing "seed" or misfolded aggregate [40]. The phenomenon extends beyond single-protein systems to cross-seeding, where aggregates of one protein type can catalyze the misfolding and aggregation of a different, heterologous protein [92] [93]. This molecular crosstalk significantly complicates neurodegenerative disease pathologies, often leading to mixed proteinopathies observed in post-mortem human brains [93]. For instance, Alzheimer's disease brains frequently exhibit both amyloid-β plaques and tau tangles, while many Parkinson's disease brains show α-synuclein-positive Lewy bodies that also contain tau [94]. The dynamic interplay between these pathological proteins accelerates disease progression and contributes to the clinical heterogeneity observed in patients [40].
Understanding and mitigating these seeding and cross-seeding processes represents a critical frontier in developing effective therapies for neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, frontotemporal dementia, and amyotrophic lateral sclerosis [40] [10]. The intricate network of protein interactions suggests that successful therapeutic strategies will likely require a multi-target approach rather than focusing on individual proteins in isolation [11]. This technical guide comprehensively examines the molecular mechanisms underlying seeding and cross-seeding, quantitative assessment methodologies, and emerging therapeutic strategies aimed at disrupting these pathological processes.
Molecular Mechanisms of Seeding and Cross-Seeding
Fundamental Seeding Mechanisms
The aggregation of prion-like proteins follows a nucleation-dependent polymerization mechanism characterized by a slow nucleation phase and a rapid elongation phase [40]. During the nucleation phase, soluble monomers undergo conformational changes to form stable, β-sheet-rich nuclei. This thermodynamically unfavorable step represents the rate-limiting barrier in the aggregation process. Once formed, these nuclei act as templates for the rapid elongation phase, where they recruit additional monomers and facilitate their conversion into the pathological conformation, leading to fibril growth [92]. Pathological proteins exhibit remarkable structural adaptability, forming distinct conformational variants known as "strains" that possess unique seeding activities, toxicities, and typical regional distributions in the brain [40]. These strain differences are now believed to underlie the clinical and pathological heterogeneity observed within and across neurodegenerative disease classifications [40].
The propagation of pathology throughout the brain occurs through cell-to-cell transmission of these protein seeds. This prion-like spreading mechanism involves the release of seeds from affected cells, their transit through the extracellular space, and their uptake by connected neurons, where they template further aggregation [10] [94]. The transmission can occur through various mechanisms, including exosomal release, tunneling nanotubes, or direct uptake of free seeds [10]. This spreading pattern often follows neuroanatomically connected pathways, explaining the predictable progression of pathology observed in many neurodegenerative diseases [94].
Cross-Seeding Dynamics and Molecular Determinants
Cross-seeding represents a sophisticated molecular interaction where pre-formed fibrils of one protein catalyze the aggregation of a different protein species. This phenomenon is not universal but depends on specific physicochemical compatibilities between the seed and recipient protein [92]. Research has demonstrated that amyloid fibrils from food proteins (lysozyme) can cross-seed α-synuclein aggregation, while fibrils from β-lactoglobulin under identical conditions cannot, highlighting the selectivity of these interactions [92].
The molecular mechanisms governing cross-seeding efficiency involve several critical factors:
- Electrostatic complementarity: The net charge and charge distribution significantly influence cross-seeding efficacy. For example, the efficient cross-seeding of α-synuclein by positively charged lysozyme fibrils is driven by electrostatic interactions with α-synuclein's negatively charged C-terminal domain, which leaves the aggregation-prone NAC domain solvent-exposed and available for nucleation [92].
- Structural compatibility: Despite lacking sequence homology, cross-seeding proteins must share structural complementarity at the β-sheet interface to facilitate templating.
- Binding orientation: The spatial orientation of the monomer on the heterologous seed surface determines whether the nucleation-competent domains remain accessible for further aggregation.
Table 1: Key Pathological Proteins Involved in Seeding and Cross-Seeding
| Protein | Primary Disease Association | Domain Critical for Aggregation | Known Cross-Seeding Partners |
|---|---|---|---|
| Amyloid-β (Aβ) | Alzheimer's Disease | Central hydrophobic cluster | Tau, α-synuclein, prion protein |
| Tau | Alzheimer's disease, Tauopathies | Microtubule-binding repeat domains | Aβ, α-synuclein |
| α-Synuclein | Parkinson's Disease, Lewy Body Dementia | NAC domain (residues 61-95) | Aβ, tau, prion protein |
| TDP-43 | ALS, FTLD | Prion-like domain | Not well characterized |
| Huntingtin | Huntington's Disease | Polyglutamine expansion | Not well characterized |
Beyond direct protein-protein interactions, the cellular environment significantly influences seeding and cross-seeding dynamics. Post-translational modifications including phosphorylation, truncation, and ubiquitination dramatically alter the aggregation propensity and seeding efficiency of pathological proteins [94]. For example, C-terminal truncation of α-synuclein significantly accelerates its aggregation by removing the protective negatively charged domain [94]. Similarly, hyperphosphorylation of tau reduces its microtubule-binding capacity and promotes its aggregation into pathological filaments [94]. The intricate balance of proteostatic mechanisms including molecular chaperones, ubiquitin-proteasome system, and autophagy pathways normally prevents pathological aggregation, but aging and cellular stress can compromise these systems, enabling seeding and cross-seeding to proceed unchecked [10] [11].
Quantitative Assessment of Seeding Phenomena
Kinetic Analysis of Seeding Efficiency
The quantitative assessment of seeding and cross-seeding efficiency primarily relies on monitoring the aggregation kinetics of the substrate protein in the presence and absence of pre-formed seeds. The thioflavin-T (ThT) fluorescence assay serves as the gold standard for these measurements, as ThT exhibits enhanced fluorescence upon binding to the cross-β-sheet structure of amyloid fibrils [92]. In this assay, the lag phase duration provides the most sensitive parameter for quantifying seeding efficiency, with effective seeds significantly reducing or eliminating this nucleation-dependent phase [92].
Table 2: Quantitative Parameters for Seeding Kinetics
| Parameter | Description | Typical Values (α-synuclein) | Interpretation |
|---|---|---|---|
| Lag Phase Duration | Time before rapid aggregation phase | 41 ± 10 hours (unseeded) | Shorter lag phase indicates higher seeding potency |
| Apparent Rate Constant (kₐₚₚ) | Slope of growth phase | Varies with conditions | Faster growth indicates efficient elongation |
| Final Fluorescence Intensity | Plateau phase ThT signal | Dependent on protein concentration | Reflects total amyloid mass formed |
| Seeding Efficiency | Ratio of seeded to unseeded lag times | 19.5 ± 1.7 hours (lysozyme-seeded) | Values <1 indicate seeding; lower values indicate stronger seeding |
The seeding efficiency can be quantified using the following relationship:
Seeding Efficiency (%) = [(tₗₐg,ᵤₙₛₑₑdₑd - tₗₐg,ₛₑₑdₑd) / tₗₐg,ᵤₙₛₑₑdₑd] × 100
Where tₗₐg,ᵤₙₛₑₑdₑd and tₗₐg,ₛₑₑdₑd represent the lag phase durations in the absence and presence of seeds, respectively. For cross-seeding experiments, it is essential to include appropriate controls to ensure that the observed effects result from genuine cross-seeding rather than non-specific aggregation enhancement [92].
Seed Amplification Assays for Diagnostic Applications
Seed amplification assays (SAAs) have emerged as powerful tools for detecting minute quantities of pathological seeds in biological fluids, offering unprecedented diagnostic sensitivity for neurodegenerative diseases [94]. These assays exploit the self-propagating nature of protein seeds by providing optimal conditions for template-directed amplification, effectively converting the detection challenge from one of quantity to one of time [94]. The real-time quaking-induced conversion (RT-QuIC) assay represents a prominent SAA variant that has been successfully applied to α-synuclein, tau, and Aβ detection in cerebrospinal fluid, plasma, and even peripheral tissues [94].
The quantitative parameters derived from SAAs include:
- Amplification lag time: The time required for the fluorescence signal to exceed a predetermined threshold, inversely correlated with seed concentration.
- Amplification rate: The slope of the growth phase, reflecting seeding efficiency.
- Maximum fluorescence: The plateau phase intensity, indicative of total amyloid formation.
These parameters not only provide diagnostic information but also enable the quantification of seeding activity in different biological samples, facilitating the stratification of patients based on their seeding burden and potency [94]. Furthermore, SAA has revealed that different diseases are associated with distinct seeding activities; for instance, cerebrospinal fluid from multiple system atrophy patients exhibits higher α-synuclein seeding activity compared to Parkinson's disease patients, suggesting the existence of disease-specific strains with varying seeding capacities [94].
Experimental Models and Methodologies
In Vitro Seeding Assays
Reductionist in vitro systems provide controlled environments for dissecting the molecular mechanisms of seeding and cross-seeding without the complexity of cellular systems.
Protocol: Fibril Preparation and Seed Generation
- Monomer Purification: Recombinantly express and purify the protein of interest (e.g., α-synuclein, tau) using ion-exchange and size-exclusion chromatography. Confirm monomeric state by analytical ultracentrifugation or size-exclusion chromatography.
- Fibrillization Reaction: Incubate monomer (50-100 μM) in aggregation buffer (e.g., for α-synuclein: 20 mM Tris-HCl, pH 7.4, 100 mM NaCl) with constant shaking (200-300 rpm) at 37°C for 3-7 days.
- Fibril Validation: Confirm fibril formation by ThT fluorescence and atomic force microscopy (AFM). Characterize morphological parameters including fibril height, periodicity, and overall structure.
- Seed Preparation: Sonicate fibrils on ice using a microtip sonicator (30-60 pulses of 1-second duration at 10-20% amplitude) to generate short fibrillar fragments. Remove insoluble material by centrifugation (15,000 × g for 10 minutes).
- Seed Quantification: Determine seed concentration based on monomer equivalents using spectrophotometry. Aliquot and store at -80°C until use [92].
Protocol: Cross-Seeding Kinetics Assay
- Reaction Setup: Combine substrate monomer (70 μM) with heterologous seeds (1-5% molar ratio of seed:monomer) in aggregation buffer in a 96-well plate. Include ThT (20 μM) for continuous monitoring.
- Kinetic Measurements: Monitor ThT fluorescence (excitation 440 nm, emission 480 nm) in a plate reader with continuous orbital shaking (200 rpm) at 37°C. Take measurements every 15-30 minutes for 48-100 hours.
- Data Analysis: Fit fluorescence versus time data to a sigmoidal curve and extract kinetic parameters (lag time, apparent growth rate, maximum fluorescence) [92].
- Morphological Analysis: Characterize resulting fibrils by AFM to determine if seed morphology was transmitted or if new fibril structures formed.
Cellular Models of Seeding and Cross-Seeding
Cell-based systems provide more physiological environments for studying seeding phenomena, including cellular uptake, subcellular localization, and proteostatic interactions.
Protocol: Cell Culture Seeding Assay
- Cell Line Selection: Utilize neuroblastoma lines (SH-SY5Y, N2a) or primary neuronal cultures depending on experimental requirements.
- Seed Transduction: Introduce pre-formed seeds (1-5 μM monomer equivalent) using lipid-based transfection reagents or add directly to culture medium. For difficult-to-transduce cells, employ protein transduction domains (e.g., TAT, Pep-1) fused to seeds.
- Incubation and Analysis: Incubate cells for 24-96 hours, then fix and immunostain for the protein of interest and aggregation markers (e.g., pS129 for α-synuclein, MC1 for pathological tau).
- Quantification: Measure the percentage of cells with inclusions, inclusion size distribution, and subcellular localization using high-content imaging systems.
- Viability Assessment: Evaluate cell health using MTT, Alamar Blue, or LDH release assays to correlate seeding with toxicity [94].
Animal Models of Seeding Propagation
In vivo models enable the study of seeding and cross-seeding in the context of intact neural circuits and complex brain environments.
Protocol: Intracerebral Seed Injection
- Seed Preparation: Generate and characterize seeds as described in section 4.1.1. Ensure endotoxin-free preparation for in vivo use.
- Stereotactic Injection: Anesthetize animals and position in stereotactic frame. Inject seeds (1-5 μL containing 1-10 μg protein) into specific brain regions (e.g., striatum, hippocampus) using a Hamilton syringe at a slow infusion rate (0.2 μL/min).
- Incubation Period: Allow pathology to develop for weeks to months, depending on the model and protein being studied.
- Tissue Analysis: Process brains for immunohistochemistry using phosphorylation-specific and conformation-specific antibodies. Quantify pathology propagation from injection site to connected regions [94].
- Behavioral Assessment: Perform motor and cognitive testing to correlate pathological spread with functional deficits.
Diagram 1: Experimental workflow for studying seeding and cross-seeding phenomena across reductionist, cellular, and animal models.
Therapeutic Strategies for Mitigating Seeding
Immunotherapeutic Approaches
Passive immunization using monoclonal antibodies represents a promising strategy for targeting pathological seeds in the extracellular space, thereby preventing their cell-to-cell transmission [94]. Several antibodies targeting α-synuclein and tau have advanced to clinical trials, with mixed results to date.
Table 3: Immunotherapeutic Approaches Against Pathological Seeds
| Target | Antibody | Development Stage | Mechanism of Action | Key Findings |
|---|---|---|---|---|
| α-Synuclein | Prasinezumab | Phase II completed | Binds aggregated α-synuclein | Slowed motor progression in rapidly progressing PD subpopulation |
| α-Synuclein | Cinpanemab | Phase II completed | Binds α-synuclein | No significant clinical benefit in overall PD population |
| Tau | Zagotenemab | Phase II completed | Binds extracellular N-terminal tau | No significant clinical benefit in early AD |
| Tau | Semorinemab | Phase II completed | Binds extracellular N-terminal tau | No significant clinical benefit in mild AD |
| Tau | Tilavonemab | Phase II completed | Binds extracellular N-terminal tau | No significant clinical benefit in PSP |
The limited clinical success of these immunotherapies highlights the challenges of targeting predominantly intracellular aggregates with antibodies that primarily access extracellular spaces [94]. However, the potential partial success of prasinezumab in specific PD subpopulations suggests that better patient stratification and earlier intervention might improve outcomes [94]. Next-generation immunotherapies are exploring antibodies that specifically target pathological conformations (strains) rather than total protein levels, which might improve specificity and reduce side effects [94].
Small Molecule Inhibitors of Seeding
Small molecules offer advantages of better blood-brain barrier penetration and intracellular activity compared to antibodies. Current strategies include:
- Seeding inhibitors: Compounds that stabilize native protein conformations or block the template surface of seeds. For example, anle138b, an oligomer modulator, has shown efficacy in multiple proteinopathy models [11].
- Strain-specific compounds: Molecules that selectively recognize and disrupt disease-specific strains while sparing physiological protein forms.
- Proteostasis enhancers: Compounds that boost cellular quality control mechanisms, including autophagy inducers (e.g, rapamycin analogs) and Hsp90 inhibitors that activate heat shock response [10] [11].
Multi-Target and Combination Therapies
Given the prevalence of mixed proteinopathies and cross-seeding phenomena, multi-target therapeutic approaches represent a promising direction [93] [11]. Strategic combinations might include:
- Sequestration agents: Molecular tweezers like CLR01 that disrupt hydrophobic interactions common to amyloid formation without specific protein targeting [11].
- Cellular clearance enhancers: Autophagy inducers (e.g., rapalogs) coupled with lysosomal activators to enhance degradation of multiple aggregated proteins [11].
- Kinase modulators: Balanced inhibition of kinases involved in pathological phosphorylation of multiple proteins (GSK3β, CK1, etc.) without completely disrupting essential signaling [11].
Diagram 2: Therapeutic strategies targeting different stages of the seeding and cross-seeding cascade.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for Seeding and Cross-Seeding Studies
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Recombinant Proteins | Human wild-type α-synuclein, Tau (2N4R, 1N4R, etc.), Aβ(1-42) | Source of monomers for aggregation studies | Ensure >95% purity and monomeric state by SEC; avoid freeze-thaw cycles |
| Pre-formed Fibrils (PFFs) | α-synuclein PFFs, Tau PFFs, Aβ PFFs | Seeds for in vitro and cellular seeding assays | Characterize by AFM/TEM; standardize sonication protocol |
| Aggregation Reporters | Thioflavin T (ThT), Proteostat dye | Monitor amyloid formation kinetics | ThT may inhibit aggregation at high concentrations; validate linear range |
| Pathological Antibodies | Anti-pS129 α-synuclein, AT8 (pS202/pT205 tau), MC1 (conformational tau) | Detect pathological species in cells and tissues | Optimize for specific applications (IHC, WB, ELISA) |
| Seed Detection Assays | RT-QuIC, PMCA | Ultrasensitive detection of seeding activity in biosamples | Strict contamination controls required; include internal standards |
| Cellular Models | SH-SY5Y, N2a, primary neurons, iPSC-derived neurons | Physiological environments for seeding studies | Characterize endogenous protein expression; optimize transduction |
| Animal Models | WT rodent models, transgenic lines (e.g., Tg2576, M83) | In vivo seeding and propagation studies | Standardize injection coordinates and seed preparations |
| Kinetic Analysis Tools | AmyloidFit, TraceDrawer | Extract kinetic parameters from aggregation data | Compare models (nucleated polymerization, F-W) for best fit |
The intricate phenomena of seeding and cross-seeding represent fundamental mechanisms driving the progression and pathological complexity of neurodegenerative diseases. While significant advances have been made in understanding the molecular principles governing these processes, several challenges remain. The development of effective mitigation strategies will require a multifaceted approach that addresses both the seed-dependent propagation and the cellular environment that enables it.
Future directions should prioritize:
- Strain-specific therapeutics that target the most pathogenic conformers while sparing physiological protein forms.
- Advanced biomarker development for early detection of seeding activity and patient stratification.
- Combination therapies that simultaneously target multiple nodes in the seeding cascade.
- Novel delivery strategies to overcome biological barriers and achieve effective intracellular concentrations of therapeutic agents.
The convergence of structural biology, biophysics, and neurobiology continues to provide unprecedented insights into the seeding and cross-seeding mechanisms. As our understanding of these processes deepens, so too will our ability to develop targeted interventions capable of slowing or halting the progression of neurodegenerative proteinopathies.
Enhancing Cellular Clearance Mechanisms to Counteract Aggregate Accumulation
Protein aggregation is a defining pathological feature of numerous neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) [16] [17]. The accumulation of misfolded protein aggregates, such as amyloid-β (Aβ) and tau in AD, and α-synuclein in PD, disrupts cellular homeostasis, impairs neuronal function, and ultimately leads to cell death [17]. Within the context of protein misfolding diseases, cellular clearance mechanisms play a critical role in maintaining proteostasis by eliminating aberrant proteins before they can form toxic oligomers and larger aggregates [16] [95]. When these systems become impaired or overwhelmed, pathological aggregates accumulate, creating a self-perpetuating cycle of neurodegeneration [96]. This technical review examines the principal pathways for aggregate clearance, provides detailed experimental methodologies for their investigation, and discusses emerging therapeutic strategies to enhance these natural defense systems, offering a scientific framework for researchers and drug development professionals targeting proteinopathies.
Core Cellular Clearance Pathways
The two major systems responsible for the clearance of aggregated proteins are the ubiquitin-proteasome system (UPS) and the lysosomal degradation pathway, which includes autophagy, endocytosis, and phagocytosis [24]. The coordinated action of these pathways is essential for preventing the accumulation of toxic protein species.
The Ubiquitin-Proteasome System (UPS)
The UPS is the primary mechanism for degrading intracellular proteins, including short-lived, damaged, and misfolded proteins [24] [97]. This process involves a highly specific enzymatic cascade:
- Ubiquitin Activation: An E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent reaction [24].
- Ubiquitin Conjugation: The activated ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme [24].
- Ubiquitin Ligation: An E3 ubiquitin ligase recognizes the target protein and facilitates the transfer of ubiquitin from E2 to the protein substrate, forming a polyubiquitin chain [24].
- Proteasomal Degradation: The polyubiquitinated protein is recognized by the 26S proteasome, which unfolds the protein, deubiquitinates it, and translocates it into the proteolytic core for degradation into small peptides and amino acids [24].
The UPS has demonstrated the capability to degrade amyloid-β peptides in a dose-dependent manner, with E2 conjugating enzymes, E3 ligases, and deubiquitinating enzymes playing pivotal roles in this process [97]. Research indicates that the UPS in glial cells is more efficient at degrading aggregated proteins compared to neurons, which may explain the relative resistance of glia to aggregate accumulation [97].
Lysosomal Degradation Pathways
Lysosomes are acidic, membrane-bound organelles containing a variety of hydrolases that degrade extracellular proteins, cell-surface receptors, and cytoplasmic components through several distinct mechanisms [24]:
- Autophagy: A critical process for degrading cytoplasmic proteins and organelles during intracellular stress or nutrient deprivation. During autophagy, a phagophore envelopes cytoplasmic components, forming a double-membrane autophagosome that subsequently fuses with a lysosome for proteolysis [24]. The autophagy-lysosomal pathway becomes activated by intracellular stress and is essential for normal growth control [24].
- Receptor-Mediated Endocytosis: Ligand binding to cell-surface receptors triggers internalization via clathrin-coated pits, forming endocytic vesicles. Acidic pH within these vesicles initiates ligand-receptor dissociation, directing contents toward lysosomal degradation [24] [97].
- Phagocytosis: Specialized cells, including microglia, engulf large particulate matter such as microorganisms or apoptotic cells, forming phagosomes that fuse with lysosomes for content degradation [24] [97].
- Pinocytosis: Non-specific engulfment of extracellular fluid and solutes, forming vesicles that fuse with lysosomes for content digestion [24].
Table 1: Major Cellular Clearance Pathways for Protein Aggregates
| Clearance Pathway | Key Components | Primary Substrates | Cellular Localization |
|---|---|---|---|
| Ubiquitin-Proteasome System (UPS) | E1-E3 enzymes, 26S proteasome | Short-lived, misfolded, and damaged intracellular proteins [24] | Cytosol, Nucleus |
| Autophagy | Phagophore, Autophagosome, Lysosomal hydrolases | Protein aggregates, Damaged organelles [24] | Cytosol |
| Receptor-Mediated Endocytosis | Cell-surface receptors, Clathrin-coated pits, Lysosomes | Extracellular ligands, Receptor-bound proteins [24] | Plasma Membrane, Endosomes |
| Phagocytosis | Phagosomes, Lysosomes | Large particles, Apoptotic cells [24] | Plasma Membrane, Phagosomes |
Specialized Mechanisms for Amyloid-β Clearance
In Alzheimer's disease, glial cells play a central role in regulating amyloid-β levels through specialized clearance mechanisms [97]. Microglia and astrocytes produce several Aβ-degrading proteases and participate in receptor-mediated clearance:
- Neprilysin (NEP): A metalloendopeptidase that degrades both monomeric and oligomeric Aβ species, primarily expressed in neurons but also found in activated astrocytes and microglia [98] [97].
- Insulin-Degrading Enzyme (IDE): A metalloendopeptidase secreted by neurons and microglia that acts on extracellular Aβ deposits, with expression patterns coinciding with insulin receptor locations in the brain [98] [97].
- Endothelin-Converting Enzymes (ECE-1 and ECE-2): Metalloproteases expressed in neurons, endothelial cells, and astrocytes that primarily degrade Aβ intracellularly [98] [97].
- Matrix Metalloproteinases (MMPs): Enzymes expressed and secreted by astrocytes that degrade both monomeric and fibrillar forms of Aβ, with MMP-2 and MMP-9 showing enhanced expression in astrocytes surrounding amyloid plaques [97].
- Receptor-Mediated Phagocytosis: Microglia and astrocytes internalize Aβ via receptors including scavenger receptors (SR), formyl peptide receptors (FPR), lipoprotein receptor-related protein 1 (LRP1), and triggering receptor expressed on myeloid cells 2 (TREM2) [97].
Table 2: Key Amyloid-β Degrading Enzymes and Their Characteristics
| Enzyme | Class | Primary Action on Aβ | Cellular Expression |
|---|---|---|---|
| Neprilysin (NEP) | Metalloendopeptidase | Degrades monomeric and oligomeric forms [97] | Neurons, Activated Astrocytes, Microglia [97] |
| Insulin-Degrading Enzyme (IDE) | Metalloendopeptidase | Degrades monomeric species [98] [97] | Neurons, Microglia, Oligodendrocytes [97] |
| Endothelin-Converting Enzyme (ECE) | Metalloendopeptidase | Intracellular degradation [98] [97] | Neurons, Endothelial cells, Astrocytes [97] |
| Matrix Metalloproteinase (MMP-2/9) | Metalloprotease | Degrades monomeric and fibrillar forms [97] | Astrocytes [97] |
| Cathepsin B | Lysosomal Peptidase | Degrades phagocytosed Aβ in microglia [97] | Microglial Lysosomes [97] |
Figure 1. Major Cellular Pathways for Protein Aggregate Clearance. The diagram illustrates the two primary degradation systems: the Ubiquitin-Proteasome System (UPS) for targeted intracellular protein degradation, and various Lysosomal Degradation Pathways including autophagy, endocytosis, and phagocytosis for bulk clearance of protein aggregates.
Experimental Protocols for Studying Clearance Mechanisms
Assessing Proteasomal Activity
Objective: To measure proteasome-mediated degradation of target proteins in cellular models.
Materials:
- MG132 (proteasome inhibitor) [96]
- Lactacystin (specific proteasome inhibitor) [96]
- Anti-ubiquitin antibodies (for immunoblotting)
- Fluorogenic proteasome substrates (e.g., Suc-LLVY-AMC for chymotrypsin-like activity)
Methodology:
- Cell Treatment: Treat cultured cells (e.g., HEK293, primary neurons) with proteasome inhibitors (e.g., 10μM MG132 or 10μM lactacystin) for 4-24 hours [96].
- Protein Extraction: Lyse cells in RIPA buffer containing protease inhibitors.
- Ubiquitin Conjugate Detection: Separate proteins by SDS-PAGE and perform immunoblotting with anti-ubiquitin antibodies to detect accumulated polyubiquitinated proteins.
- Proteasome Activity Assay: Incubate cell lysates with fluorogenic substrates (100μM Suc-LLVY-AMC) in assay buffer (50mM Tris-HCl, pH 7.5, 5mM MgCl₂, 1mM DTT) for 1 hour at 37°C.
- Quantification: Measure fluorescence (excitation 380nm, emission 460nm) and normalize to protein concentration. Compare inhibitor-treated samples to vehicle controls.
Data Interpretation: Proteasome inhibition should increase polyubiquitinated protein levels and decrease proteolytic activity. A 50-80% reduction in activity is typically observed with effective inhibition.
Monitoring Autophagic Flux
Objective: To quantitatively measure autophagic activity and lysosomal degradation.
Materials:
- Bafilomycin A1 (V-ATPase inhibitor that blocks autophagosome-lysosome fusion)
- Chloroquine (lysosomotropic agent that inhibits autophagic degradation)
- Anti-LC3B antibody (marker for autophagosomes)
- Anti-p62/SQSTM1 antibody (selective autophagy substrate)
- LC3-GFP plasmid (for live-cell imaging of autophagosomes)
Methodology:
- Treatment: Incubate cells with 100nM bafilomycin A1 or 50μM chloroquine for 4-6 hours to block lysosomal degradation.
- Immunoblotting: Extract proteins and perform Western blotting for LC3 and p62.
- LC3 Conversion: Monitor LC3-I to LC3-II conversion; increased LC3-II indicates autophagosome accumulation.
- p62 Degradation: Decreased p62 levels indicate functional autophagic flux.
- Microscopy: For live-cell imaging, transfect cells with LC3-GFP and count puncta per cell before and after treatment.
Data Interpretation: Functional autophagy shows increased LC3-II and decreased p62 in basal conditions. With lysosomal inhibitors, further accumulation of LC3-II and p62 indicates active autophagic flux.
Evaluating Aβ Phagocytosis by Glial Cells
Objective: To quantify the uptake and degradation of Aβ by microglia and astrocytes.
Materials:
- Fluorescently-labeled Aβ(1-42) (e.g., FITC-Aβ)
- Primary microglial or astrocyte cultures
- Protease inhibitors (e.g., protease inhibitor cocktail)
- Aβ-degrading enzyme inhibitors (e.g., thiorphan for NEP, insulin for IDE)
Methodology:
- Preparation: Reconstitute FITC-Aβ(1-42) in PBS to 100μM and aggregate by incubating at 37°C for 24 hours.
- Treatment: Add 1μM FITC-Aβ to glial cultures for 2-24 hours.
- Inhibition Studies: Pre-treat cells with enzyme inhibitors (10μM thiorphan for NEP, 100nM insulin for IDE) for 1 hour before Aβ addition.
- Quantification:
- Flow Cytometry: Trypsinize cells and analyze FITC fluorescence by flow cytometry.
- Immunofluorescence: Fix cells and counterstain with DAPI; quantify fluorescence intensity per cell.
- ELISA: Measure remaining Aβ in medium using human Aβ ELISA kits.
Data Interpretation: Effective phagocytosis shows time-dependent increase in intracellular fluorescence. Inhibitor treatment should decrease Aβ degradation and increase extracellular Aβ levels.
Therapeutic Strategies to Enhance Clearance
Emerging therapeutic approaches aim to boost cellular clearance mechanisms to counteract aggregate accumulation in neurodegenerative diseases.
Pharmacological Enhancement of Proteostasis
Small molecule approaches target various components of the protein quality control system:
- HSP70 Inducers: Compounds that enhance heat shock protein 70 (HSP70) transcription have been shown to promote TDP-43 aggregate clearance and improve cell viability in ALS models [96].
- Autophagy Enhancers: Rapamycin and its analogs (rapalogs) induce autophagy by inhibiting mTOR signaling, promoting the clearance of various protein aggregates including mutant huntingtin and α-synuclein [24].
- Proteasome Activators: Small molecules that enhance proteasomal activity, such as polyphenols and other natural compounds, show potential for increasing clearance of ubiquitinated proteins [24].
Immunotherapeutic Approaches
Antibody-based strategies have shown promise in selectively targeting pathological protein aggregates:
- α-miSOD1: A recombinant human monoclonal antibody that selectively binds misfolded SOD1 without targeting physiological SOD1 dimers. In transgenic ALS mice expressing human SOD1 mutations, α-miSOD1 treatment delayed motor symptom onset, extended survival by up to 2 months, and reduced aggregation of misfolded SOD1 and motor neuron degeneration [96].
- Single-Chain Variable Fragments (scFvs): Engineered antibody fragments targeting specific epitopes on aggregated proteins. The 3B12A scFv, which recognizes a region of TDP-43 nuclear export signal, accelerated proteasome-mediated degradation of aggregated TDP-43 and reduced aggregates in embryonic mouse brain without apparent side effects [96]. Similarly, the VH7Vk9 scFv against the RRM1 domain of TDP-43 reduced the cytoplasmic/nuclear TDP-43 ratio and improved clearance via proteasome and autophagosomes [96].
Targeted Protein Degradation Technologies
Novel approaches that hijack natural degradation systems show significant potential:
- PROteolysis TArgeting Chimeras (PROTACs): Heterobifunctional molecules with ligands specific for both a protein of interest (POI) and an E3 ubiquitin ligase. PROTACs simultaneously bind the POI and E3 ligase, inducing ubiquitylation and subsequent degradation by the UPS. This technology enables targeted degradation of intracellular proteins that have been difficult to manipulate with traditional small molecules [24].
- LYsosome-TArgeting Chimeras (LYTACs): Bifunctional molecules that bind simultaneously to endogenous cell-surface lysosome-targeting receptors and extracellular proteins. Upon engagement, the target protein is internalized by clathrin-mediated endocytosis and transported to the lysosome for degradation, enabling targeting of extracellular proteins including secreted and membrane-bound proteins [24].
Gene Therapy Approaches
Gene-based strategies aim to enhance the expression of key clearance components:
- NEP and IDE Overexpression: Gene delivery of Aβ-degrading enzymes such as neprilysin and insulin-degrading enzyme has been shown to reduce Aβ load in animal models, providing proof-of-concept for enzyme augmentation strategies [98] [97].
- Antisense Therapies: Antisense oligonucleotides that prevent cells from making specific death fold proteins are being explored by biotech companies as potential treatments for Alzheimer's and other neurodegenerative diseases [99].
Table 3: Experimental Models for Studying Aggregate Clearance
| Experimental Model | Key Applications | Advantages | Limitations |
|---|---|---|---|
| Primary Neuronal Cultures | Study cell-autonomous clearance mechanisms [96] | Physiologically relevant, Responsive to stressors | Limited lifespan, Neuronal heterogeneity |
| Glial Cell Cultures (Microglia, Astrocytes) | Investigate Aβ phagocytosis, Inflammatory responses [97] | Primary mediators of clearance, Can be activated | May differ from in vivo phenotypes |
| HEK293 Cell Line | Protein aggregation, Transfection studies [96] | High transfection efficiency, Rapid growth | Non-neuronal origin, Limited relevance |
| Transgenic Mouse Models (APP/PS1, SOD1) | In vivo clearance, Therapeutic testing [96] [97] | Complex tissue environment, Disease progression | Species differences, Costly and time-consuming |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Studying Protein Aggregate Clearance
| Reagent/Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Lactacystin [96] | Block proteasomal activity to assess UPS function | Use at 10-20μM for 4-24 hours; monitor ubiquitin conjugates |
| Lysosomal Inhibitors | Bafilomycin A1, Chloroquine [24] | Inhibit lysosomal degradation to measure autophagic flux | Bafilomycin A1 (100nM) more specific; Chloroquine (50μM) cost-effective |
| Aβ-Degrading Enzyme Inhibitors | Thiorphan (NEP inhibitor) [97], Insulin (IDE competitor) [97] | Block specific Aβ degradation pathways | Use to identify contribution of specific enzymes to Aβ clearance |
| Autophagy Markers | LC3B Antibodies, p62/SQSTM1 Antibodies [24] | Detect autophagosomes and monitor autophagic flux | LC3-II:P62 ratio indicates autophagic activity; combine with inhibitors |
| Fluorescent Protein Aggregates | FITC-Aβ(1-42) [97], Htt-Q74-GFP | Track phagocytosis and intracellular fate | Pre-aggregate for 24-48h at 37°C for consistent results |
| Lysosomal Probes | pHrodo pH indicators, LysoTracker [24] | Monitor lysosomal pH and localization | pHrodo increases fluorescence in acidic environments; useful for endocytosis |
| PROTAC Molecules | Heterobifunctional degraders [24] | Induce targeted protein degradation via UPS | Requires target ligand and E3 ligase ligand; cell-permeable designs |
| Cellular Stress Inducers | Rapamycin (autophagy inducer) [24], Proteotoxic stressors | Activate clearance pathways | Rapamycin (100-200nM) induces autophagy via mTOR inhibition |
Figure 2. Experimental Workflow for Clearance Mechanism Analysis. This diagram outlines a systematic approach for investigating cellular clearance pathways, from model selection through therapeutic treatment and pathway inhibition to comprehensive assessment using multiple analytical methods.
Enhancing cellular clearance mechanisms represents a promising therapeutic strategy for counteracting protein aggregate accumulation in neurodegenerative diseases. The ubiquitin-proteasome and lysosomal degradation pathways provide complementary systems for maintaining proteostasis, while specialized mechanisms in glial cells offer additional targets for intervention. Experimental approaches combining pharmacological tools, genetic manipulations, and advanced imaging techniques continue to reveal new insights into the regulation of these clearance systems. Emerging technologies such as PROTACs and LYTACs demonstrate the potential of harnessing natural degradation machinery for targeted protein removal. As our understanding of the complex interplay between different clearance mechanisms deepens, so too will opportunities to develop effective therapies that can break the cycle of protein aggregation and neurodegeneration.
The development of effective biotherapeutics for neurodegenerative diseases represents one of the most challenging frontiers in pharmaceutical science. These diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Amyotrophic Lateral Sclerosis (ALS), are characterized by the pathological aggregation of specific proteins [100] [40]. In AD, amyloid-β (Aβ) peptides aggregate to form amyloid plaques, while hyperphosphorylated tau protein forms neurofibrillary tangles [100]. Similarly, PD is characterized by Lewy bodies formed by aggregated α-synuclein protein [100] [40]. The intrinsically disordered nature of many of these proteins, such as Aβ, tau, and α-synuclein, challenges conventional drug discovery paradigms that typically target well-defined binding pockets [101]. Protein aggregation is not merely a pathological endpoint but a dynamic process driven by multiple molecular mechanisms including proteolytic cleavage, point mutations, and post-translational modifications that disrupt normal protein folding and stability [100]. For biotherapeutic development, aggregation poses a dual challenge: it is both a primary therapeutic target and a critical quality attribute that must be controlled during drug development to ensure product safety and efficacy [100] [102].
Table 1: Key Pathological Proteins in Neurodegenerative Diseases
| Disease | Aggregating Protein | Aggregate Structure | Cellular Location |
|---|---|---|---|
| Alzheimer's Disease (AD) | Amyloid-β (Aβ) and tau | Plaques and neurofibrillary tangles | Extracellular and intracellular |
| Parkinson's Disease (PD) | α-synuclein | Lewy bodies | Intracellular |
| Huntington's Disease (HD) | Huntingtin | Inclusion bodies | Intracellular |
| Amyotrophic Lateral Sclerosis (ALS) | TDP-43, FUS | Cytoplasmic inclusions | Intracellular |
Computational Prediction of Aggregation-Prone Regions
Fundamental Principles and Databases
Computational prediction of protein aggregation begins with understanding the fundamental driving forces behind this process. The reduction in free surface energy by the removal of hydrophobic residues from contact with the solvent is a major driving force in protein aggregation [100] [102]. Protein aggregation often follows a nucleation-growth mechanism, where an initial lag phase involves the formation of a critical nucleus, followed by rapid growth into insoluble aggregates [102]. Modern computational approaches leverage this understanding through two primary methodological frameworks: sequence-based methods that analyze amino acid patterns and physicochemical properties, and structure-based methods that incorporate three-dimensional structural information [102]. The development of these methods has been facilitated by specialized databases that curate experimental data on protein aggregation.
Table 2: Key Databases for Protein Aggregation Research
| Database Name | Primary Focus | Key Features | Applications |
|---|---|---|---|
| AmyLoad | Amyloidogenic fragments | Collects amyloidogenic and non-amyloidogenic sequence fragments | Reference data for aggregation propensity studies |
| CPAD 2.0 (Curated Protein Aggregation Database) | Experimental aggregation data | Comprehensive collection of experimental studies on aggregation | Benchmarking computational predictions |
| A3D (Aggrescan3D) | Structure-based aggregation propensity | Uses 3D models to compute aggregation scores for amino acids | Analyzing effects of mutations on solubility |
| WALTZ-DB 2.0 | Amyloid-forming peptides | Experimentally validated amyloid-forming hexapeptides | Identifying short aggregation-prone motifs |
| ProADD | Protein aggregation diseases | Classifies diseases based on sequence and structural analysis | Pattern mining of aggregating proteins |
Computational Methods and Tools
The computational toolbox for predicting protein aggregation has expanded significantly, with methods ranging from simple propensity scales to sophisticated machine learning algorithms. These tools can predict aggregation-prone regions (APRs), aggregation kinetics, and the impact of mutations on aggregation behavior [102] [42]. Tools like AGGRESCAN use aggregation propensity scales derived from in vivo experiments, while Zyggregator incorporates multiple factors including α-helix and β-sheet formation propensity, hydrophobicity, charge, and the presence of "Gatekeeper" residues that protect against aggregation [102]. TANGO employs empirical and statistically derived energy functions to predict segmental β-sheet probability, and PASTA evaluates the stability of putative cross-β pairings between different sequence stretches [102]. Recent advances include structure-based methods like Aggrescan3D (A3D), which computes structurally corrected aggregation values using 3D atomic models, enabling researchers to visualize aggregation "hot spots" on protein structures and test the effects of mutations [102].
Diagram 1: Computational Prediction Workflow
Experimental Validation of Computational Predictions
Biophysical Characterization of Protein Aggregation
Computational predictions of protein aggregation must be rigorously validated through experimental methods that characterize different aspects of the aggregation process. These techniques provide quantitative and qualitative data on aggregation kinetics, structural transitions, and morphological features. The experimental toolbox includes spectroscopic, microscopic, and calorimetric methods that collectively provide a comprehensive picture of aggregation behavior [101]. Spectroscopic techniques like Thioflavin T (ThT) fluorescence and Congo Red binding are widely used to monitor the formation of amyloid fibrils through their specific binding to cross-β-sheet structures [101]. Fourier Transform Infrared (FTIR) spectroscopy and Circular Dichroism (CD) spectroscopy provide information about secondary structural changes during aggregation, particularly the transition from disordered or α-helical structures to β-sheet-rich conformations [103] [101]. Microscopic techniques including Transmission Electron Microscopy (TEM), Atomic Force Microscopy (AFM), and Cryogenic Electron Microscopy (cryo-EM) enable direct visualization of aggregate morphology, from oligomeric species to mature fibrils [101]. Cryo-EM has been particularly transformative, providing atomic-level structures of amyloid fibrils that have revolutionized our understanding of their molecular architecture [101].
Table 3: Experimental Methods for Protein Aggregation Analysis
| Method Category | Specific Techniques | Key Measured Parameters | Applications in Formulation |
|---|---|---|---|
| Spectroscopy | Thioflavin T fluorescence, CD, FTIR | β-sheet content, aggregation kinetics, structural transitions | Screening formulation conditions, stability assessment |
| Microscopy | TEM, AFM, cryo-EM | Aggregate morphology, size distribution, fibril structure | Characterizing aggregate structures, quality control |
| Calorimetry | ITC, DSC | Binding affinity, thermal stability, aggregation energetics | Excipient selection, stability optimization |
| Size Analysis | DLS, SEC | Hydrodynamic size, oligomer distribution | Assessing colloidal stability, aggregation state |
| Binding Assays | SPR, BLI | Binding kinetics, affinity for inhibitors or antibodies | Candidate screening, epitope characterization |
Structural Biology and Advanced Characterization
For the structural characterization of protein aggregates, particularly those relevant to neurodegenerative diseases, advanced structural biology techniques are essential. Cryo-EM has emerged as a powerful method for determining the atomic structures of amyloid fibrils, revealing how different proteins adopt the characteristic cross-β architecture while maintaining structural polymorphisms that may correlate with different disease strains [101]. Solid-state Nuclear Magnetic Resonance (ssNMR) spectroscopy provides complementary information about the structural organization of amyloid fibrils, including details about side-chain packing and the dynamic properties of different regions within the aggregates [103]. X-ray crystallography has provided high-resolution structures of short amyloidogenic peptides, revealing the "steric zipper" motif formed by two self-complementary β-sheets that constitute the spine of amyloid fibrils [102] [101]. These structural insights are crucial for rational drug design, as they enable the identification of specific structural vulnerabilities that can be targeted by therapeutic interventions. For formulation scientists, understanding the structural basis of aggregation informs the design of strategies to stabilize the native state or disrupt specific interactions that lead to aggregation.
Integrated Workflow: From Prediction to Validation
The Data-Driven Formulation Cycle
The integration of computational predictions and experimental validation follows an iterative cycle that progressively refines formulation design. This data-driven workflow begins with computational screening to identify potential aggregation-prone regions in the therapeutic protein [100] [102]. These predictions inform the design of experimental studies that test specific hypotheses about aggregation behavior under different formulation conditions. The experimental results then feed back into computational models to improve their predictive accuracy, creating a virtuous cycle of knowledge generation and application. This approach is particularly valuable for optimizing therapeutic antibodies and other protein biologics targeting neurodegenerative diseases, where controlling aggregation is critical for both product quality and therapeutic efficacy [100] [104]. The ProMIS Neurosciences EpiSelect platform exemplifies this integrated approach, using computational methods to identify conformational epitopes uniquely exposed on toxic misfolded proteins, which are then validated experimentally to generate misfolding-specific antibodies [104].
Diagram 2: Data-Driven Formulation Development Cycle
Case Study: PMN310 for Alzheimer's Disease
The development of PMN310, a humanized monoclonal antibody targeting toxic amyloid-β oligomers (AβO) for Alzheimer's disease, illustrates the successful application of data-driven formulation development [104]. ProMIS Neurosciences used their computational EpiSelect platform to identify conformational epitopes uniquely exposed on toxic Aβ oligomers without significant reactivity with amyloid-beta monomers or fibrils [104]. This computational prediction enabled the generation of an antibody candidate that avoids "target distraction" by more abundant non-toxic species and potentially reduces the risk of adverse effects like amyloid-related imaging abnormalities (ARIA) associated with other amyloid-targeting therapies [104]. The PRECISE-AD Phase 1b clinical trial for PMN310 has incorporated extensive biomarker analysis to validate the computational predictions and establish proof of mechanism [104]. This case study demonstrates how computational predictions can guide the development of therapeutics with improved safety profiles by selectively targeting specific aggregation states most relevant to disease pathogenesis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful data-driven formulation development requires a comprehensive toolkit of research reagents and analytical technologies. These tools enable researchers to implement the integrated computational and experimental workflows necessary to address protein aggregation challenges.
Table 4: Essential Research Reagent Solutions for Aggregation Studies
| Reagent/Material Category | Specific Examples | Function in Formulation Development | Key Applications |
|---|---|---|---|
| Aggregation Detection Dyes | Thioflavin T, Congo Red, ANS | Selective binding to amyloid structures and hydrophobic surfaces | Monitoring aggregation kinetics, screening inhibitors |
| Stabilizing Excipients | Sugars (sucrose, trehalose), surfactants (Polysorbate 20/80) | Prevents aggregation via preferential exclusion, surface protection | Formulation optimization, stability enhancement |
| Structural Biology Reagents | Cryo-EM grids, negative stains, NMR isotopes | Enable high-resolution structural analysis of aggregates | Determining aggregate structure, mechanism elucidation |
| Binding Assay Systems | SPR chips, BLI biosensors, ITC reagents | Quantify interactions between proteins and inhibitors/excipients | Candidate screening, affinity measurements |
| Chromatography Materials | Size exclusion columns, ion exchange resins | Separate and analyze different aggregation states | Purity assessment, aggregate quantification |
Data-driven formulation development represents a paradigm shift in the creation of biotherapeutics for neurodegenerative diseases. By integrating computational predictions with rigorous experimental validation, researchers can address the complex challenge of protein aggregation more systematically and effectively. The continuing advancement of computational methods, particularly through artificial intelligence and machine learning, promises to further enhance our ability to predict aggregation behavior and design optimized formulations [67] [42]. Similarly, innovations in experimental techniques, especially in structural biology and high-throughput screening, will provide richer datasets to refine computational models [101]. The growing understanding of protein aggregation in neurodegenerative diseases, coupled with these technological advances, creates an exciting landscape for the development of next-generation therapeutics that can effectively target the toxic protein species driving these devastating conditions while maintaining the stability and efficacy required for successful clinical application.
Therapeutic Validation: Comparative Analysis of Clinical and Preclinical Strategies
Protein misfolding and aggregation are fundamental pathological hallmarks of a wide range of neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD). These conditions are characterized by the progressive accumulation of toxic protein species—such as amyloid-β (Aβ), tau, α-synuclein, and huntingtin—which disrupt cellular homeostasis and trigger cascades of neuronal dysfunction and death [105] [9]. The delicate balance between protein synthesis, folding, and degradation—collectively known as proteostasis—becomes compromised in these disorders, leading to a self-propagating cycle of protein aggregation and cellular damage [1].
Within this challenging landscape, small molecule modulators (SmMs) have emerged as a promising therapeutic class due to their unique ability to intervene at multiple stages of the aggregation cascade. Unlike larger biologic therapeutics, small molecules offer the advantage of favorable pharmacokinetics, including blood-brain barrier penetration, oral bioavailability, and tunable chemical properties [105]. These compounds can modulate protein aggregation through diverse mechanisms, including: stabilization of native protein conformations, inhibition of oligomerization and fibrillization, disruption of pre-formed fibrils, and enhancement of cellular clearance pathways [105] [106]. This comprehensive review examines the current landscape of small molecule therapeutics targeting protein misfolding and aggregation, with a specific focus on their mechanisms, experimental characterization, and clinical translation.
Molecular Mechanisms of Small Molecule Intervention
Small molecules employ sophisticated structural strategies to combat protein aggregation, leveraging precise molecular interactions to stabilize functional protein conformations or disrupt pathological assemblies. The following table summarizes the primary mechanisms through which these compounds exert their effects.
Table 1: Primary Mechanisms of Small Molecule Inhibitors of Protein Aggregation
| Mechanism of Action | Molecular Targets | Representative Agents | Biological Effect |
|---|---|---|---|
| Stabilization of Native State | Transthyretin (TTR) tetramer [106] | Tafamidis [106] | Prevents tetramer dissociation and subsequent amyloidogenesis |
| Inhibition of Oligomerization | Aβ42 protofibrils, α-synuclein oligomers [106] [107] | Lecanemab (mAb), Valiltramiprosate (ALZ-801) [107] | Blocks formation of soluble neurotoxic oligomers |
| Disruption of Pre-formed Fibrils | Mature amyloid fibrils of Aβ and α-synuclein [105] | EGCG, Kaempferol [105] [108] | Binds to β-sheet-rich structures promoting fibril disassembly |
| Tau Aggregation Inhibition | Microtubule-associated protein tau [107] | Hydromethylthionine Mesylate (HMTM) [107] | Prevents tau self-assembly into neurofibrillary tangles |
| Proteostasis Network Enhancement | Sigma-1 receptor (SIGMAR1) [107] | Blarcamesine (ANAVEX 2-73) [107] | Activates cellular clearance pathways including autophagy |
The aggregation process typically follows a nucleation-dependent polymerization mechanism, characterized by a lag phase (nucleation), exponential growth (elongation), and a final plateau phase [109]. Small molecules can intervene at any point along this pathway. For structured precursor proteins like transthyretin, kinetic stabilizers such as tafamidis bind to the native tetrameric structure, increasing the activation barrier for dissociation into aggregation-prone monomers [106]. For intrinsically disordered proteins (e.g., Aβ, α-synuclein), inhibitors often operate through β-sheet disruption or specific oligomer sequestration [105] [106].
Notably, many plant-derived polyphenols—such as epigallocatechin gallate (EGCG), genistein, and apigenin—demonstrate multi-modal inhibition, simultaneously preventing initial misfolding, stabilizing non-toxic oligomers, and promoting the dissolution of mature fibrils [105] [108]. The therapeutic precision of these compounds has been further enhanced through nano-conjugation strategies, which improve their bioavailability, target specificity, and cellular uptake [105].
Diagram 1: Small Molecule Intervention Points in the Protein Aggregation Cascade. Small molecule inhibitors (blue) target specific stages of the pathological aggregation pathway, from initial misfolding to fibril formation and clearance.
Key Pharmacological Agents and Their Targets
The landscape of small molecule inhibitors encompasses both natural compounds and synthetically designed drugs, each with distinctive structural properties and molecular targets. The following table provides a comprehensive overview of prominent agents currently under investigation.
Table 2: Key Small Molecule Agents Targeting Protein Misfolding and Aggregation
| Agent Name | Chemical Class | Primary Target | Mechanistic Action | Development Stage |
|---|---|---|---|---|
| Tafamidis [106] | Benzoxazole derivative | Transthyretin (TTR) | Tetramer stabilization, prevents dissociation | Marketed (ATTR amyloidosis) |
| Valiltramiprosate (ALZ-801) [107] | Small molecule | Amyloid-β (Aβ) | Blocks formation of neurotoxic soluble Aβ oligomers | Phase 3 (APOE4/4 carriers) |
| Hydromethylthionine Mesylate (HMTM) [107] | Tau aggregation inhibitor | Microtubule-associated protein tau | Inhibits tau self-assembly into paired helical filaments | Phase 3 (MAA submitted) |
| Blarcamesine (ANAVEX 2-73) [107] | Sigma-1 receptor agonist | Sigma-1 receptor (SIGMAR1) | Activates autophagy, restores proteostasis | Phase 2b/3 |
| EGCG [105] [108] | Polyphenol (flavonoid) | Aβ, α-synuclein | Remodels mature fibrils, generates non-toxic aggregates | Preclinical/Clinical trials |
| Genistein [105] | Isoflavone | Human islet amyloid polypeptide (hIAPP) | Dual inhibitor of hIAPP aggregation | Preclinical |
| Kaempferol [105] | Polyphenol (flavonoid) | α-Synuclein | Inhibits α-Syn aggregation in mouse models | Preclinical |
Tafamidis represents a landmark achievement in conformational therapeutics as the first approved drug specifically designed to stabilize a native protein fold against amyloidogenic dissociation. It functions by binding with high affinity to the thyroxine-binding sites of the TTR tetramer, dramatically slowing its dissociation into monomeric intermediates that readily misfold into amyloid fibrils [106].
In Alzheimer's disease, Valiltramiprosate (ALZ-801) exemplifies a precision medicine approach targeting the earliest toxic species in the aggregation cascade. This oral prodrug selectively inhibits the formation of soluble Aβ oligomers, which are now widely recognized as the primary neurotoxic agents driving synaptic dysfunction and cognitive decline. Recent Phase 3 trials specifically enrolled genetically defined populations (APOE4/4 homozygotes), revealing a 52% reduction in cognitive decline in patients with mild cognitive impairment (MCI) [107].
The tau aggregation inhibitor Hydromethylthionine Mesylate (HMTM) offers an alternative therapeutic strategy for Alzheimer's by targeting the other key pathological protein. In the Phase III LUCIDITY trial, HMTM demonstrated sustained clinical benefits over 18 months, including slowed cognitive decline and reduced brain atrophy, with a favorable safety profile across more than 3,000 participants [107].
Natural products continue to provide valuable chemical scaffolds for inhibition, with EGCG (epigallocatechin gallate) representing one of the most extensively studied polyphenols. EGCG exhibits remarkable multi-target activity, capable of redirecting amyloidogenic proteins into stable, non-toxic oligomers while also promoting the disassembly of mature fibrils through direct binding to β-sheet structures [105] [108]. Similarly, genistein and apigenin demonstrate structure-dependent inhibition, with their specific hydroxylation patterns enabling selective interference with the aggregation pathways of different amyloidogenic proteins [105] [108].
Experimental Methodologies for Evaluation
In Vitro Aggregation Kinetics Assessment
The quantitative evaluation of small molecule inhibitors relies heavily on biophysical techniques that monitor the time-dependent progression of protein aggregation. Thioflavin T (ThT) fluorescence assays represent the gold standard for tracking amyloid formation kinetics in real-time [106]. This method capitalizes on the enhanced fluorescence emission of ThT when bound to β-sheet-rich structures, providing a direct measurement of fibril formation.
Protocol for ThT Aggregation Inhibition Assay:
- Prepare amyloidogenic protein (e.g., Aβ42, α-synuclein) in aggregation-prone buffer conditions (e.g., PBS with gentle agitation).
- Pre-incubate test compound with protein monomer at varying molar ratios (typically 1:1 to 1:10 compound:protein).
- Add ThT to a final concentration of 10-20 μM and transfer to black-walled 96-well plates.
- Monitor fluorescence (excitation 440 nm, emission 480 nm) in a plate reader with continuous orbital shaking at 37°C.
- Analyze kinetic parameters: lag time duration, aggregation rate (slope of exponential phase), and final fluorescence intensity [106].
The resulting sigmoidal curve provides critical quantitative parameters for evaluating inhibitor potency. Effective compounds typically prolong the lag phase and reduce the maximum fluorescence amplitude, indicating delayed nucleation and diminished fibril yield, respectively [109].
Oligomer-Specific Toxicity Profiling
Since soluble oligomeric intermediates are now recognized as the primary cytotoxic species in protein aggregation diseases, specific methodologies have been developed to assess protection against oligomer-mediated toxicity.
Protocol for Oligomer Toxicity Assessment:
- Prepare oligomer-enriched fractions from amyloidogenic proteins using size-exclusion chromatography or controlled incubation conditions.
- Pre-treat neuronal cell lines (e.g., SH-SY5Y, PC12) or primary neurons with test compounds for 24 hours.
- Apply oligomer preparations to cultured neurons for 24-48 hours.
- Assess cell viability using MTT or WST-1 assays, and measure caspase activity as an apoptosis indicator.
- Evaluate synaptic integrity through immunocytochemistry for pre- and post-synaptic markers (e.g., synaptophysin, PSD-95) [106] [9].
Compounds like valiltramiprosate demonstrate exceptional efficacy in this paradigm, showing significant protection against oligomer-induced synaptic toxicity even at nanomolar concentrations [107].
Structural Biology and Binding Analyses
Understanding the structural basis of inhibitor action provides critical insights for rational drug design. Nuclear magnetic resonance (NMR) spectroscopy, particularly (^{15})N-(^{1})H HSQC experiments, can map compound-binding sites on amyloidogenic proteins at atomic resolution [106]. X-ray crystallography of inhibitor-protein complexes has revealed precise molecular interactions, as demonstrated in the structure of transthyretin bound to tafamidis [106]. Additionally, native mass spectrometry enables quantitative assessment of tetramer stabilization for proteins like transthyretin, providing a direct readout of thermodynamic stabilization [106].
The Scientist's Toolkit: Essential Research Reagents
Advancing research on small molecule inhibitors requires a carefully selected array of reagents and methodologies. The following table outlines essential tools for investigating protein aggregation and therapeutic intervention.
Table 3: Essential Research Reagents for Protein Aggregation Studies
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Amyloidogenic Proteins | Recombinant Aβ42, α-synuclein, tau [106] | Substrate for aggregation assays | Ensure monomer purification to prevent seeded aggregation |
| Fluorescent Dyes | Thioflavin T (ThT), ANS [106] | Fibril detection & kinetics | ThT specifically recognizes cross-β structure; ANS binds hydrophobic patches |
| Cell-Based Models | SH-SY5Y neuroblastoma, primary cortical neurons [9] | Toxicity & protection assays | Use oligomer-enriched preparations for physiological relevance |
| Protein Stability Assays | Thermal shift, SUB-EX [106] | Native state stabilization screening | Monitor Tm changes for stabilizers like tafamidis |
| Antibody Tools | Anti-oligomer A11, OC conformers [110] | Specific oligomer detection | Distinguish different aggregation states in cell lysates/tissues |
| Proteostasis Modulators | HSP70/90 inhibitors, autophagy inducers [111] [1] | Network-level mechanistic studies | Modulate cellular quality control pathways |
Clinical Translation and Current Landscape
The translation of small molecule inhibitors from preclinical models to clinical applications has accelerated dramatically in recent years, with several promising candidates advancing through late-stage trials. The field is increasingly embracing precision medicine approaches, targeting specific patient populations based on genetic biomarkers and early disease stages [107].
Lecanemab (LEQEMBI), an antibody therapeutic targeting Aβ protofibrils, has paved the way for anti-aggregation therapies with its full regulatory approval in the US. Its clinical success validates the approach of targeting soluble oligomeric species rather than insoluble plaques [107]. Small molecules like valiltramiprosate are building on this foundation by offering oral administration with a potentially improved safety profile, specifically avoiding ARIA (Amyloid-Related Imaging Abnormalities) associated with some antibody therapies [107].
The clinical development of blarcamesine highlights the emerging strategy of targeting the proteostasis network rather than individual aggregated proteins. By activating the sigma-1 receptor, blarcamesine enhances autophagy and cellular stress responses, providing a broader protective effect across multiple neurodegenerative pathways. Long-term extension studies have demonstrated sustained cognitive benefits over four years of continuous treatment, suggesting true disease-modifying potential [107].
Recent analyses indicate a robust pipeline of approximately 60 molecules in various phases of clinical development for Alzheimer's disease alone, with about 25% in Phase III trials and potentially nearing regulatory review [107]. Consortium efforts like the Global Neurodegeneration Proteomics Consortium (GNPC) are further accelerating therapeutic development by providing large-scale proteomic datasets to identify novel biomarkers and therapeutic targets [7].
Diagram 2: Therapeutic Development Pipeline for Small Molecule Aggregation Inhibitors. The pathway from preclinical discovery to clinical application involves rigorous validation and increasingly incorporates biomarker-based patient stratification strategies.
Challenges and Future Perspectives
Despite considerable progress, significant challenges remain in the development of small molecule therapies for protein misfolding disorders. Bioavailability and blood-brain barrier penetration continue to present substantial hurdles, particularly for natural polyphenols like EGCG and curcumin, which often exhibit poor pharmacokinetic profiles [105]. Innovative formulation strategies, including nano-conjugation approaches, show promise in enhancing the delivery and precision of these compounds [105]. Nano-conjugated small molecules demonstrate improved stability, targeted delivery to specific tissues, and the ability to function at nanomolar rather than millimolar concentrations [105].
The multifactorial nature of neurodegenerative diseases also complicates therapeutic intervention, as multiple pathological proteins often co-exist and potentially interact through cross-seeding mechanisms [9]. Future therapeutic strategies may require combination approaches or multi-target ligands capable of simultaneously addressing different aspects of the pathological cascade.
The ongoing revolution in structural biology and artificial intelligence is poised to dramatically accelerate small molecule discovery. Recent breakthroughs in AI-powered protein structure prediction are enabling more rational design of conformational therapeutics, while high-throughput screening technologies allow rapid identification of novel chemotypes [109] [1]. As our understanding of the structural principles governing protein misfolding deepens, so too will our ability to design precision therapeutics that can restore proteostasis and modify the progression of these devastating disorders.
The convergence of these technological advances with large-scale collaborative efforts like the GNPC—which has established one of the world's largest harmonized proteomic datasets—heralds a new era of biomarker-driven therapeutic development [7]. By integrating molecular insights with clinical applications, the next generation of small molecule inhibitors promises to fundamentally transform our approach to treating protein misfolding diseases.
Neurodegenerative diseases (NDDs) represent one of the most significant challenges in modern medicine, characterized pathologically by the accumulation of misfolded protein aggregates in the central nervous system. The fundamental mechanism underlying these conditions involves the transformation of soluble, natively folded proteins or peptides into abnormal conformations dominated by β-sheet secondary structures, which subsequently oligomerize into pathological species that exhibit both toxicity and prion-like spreading capabilities [8] [112]. This aggregation process follows a predictable pathway beginning with protein misfolding, proceeding through oligomerization, and culminating in the formation of protofibrils and mature fibrils that often deposit as insoluble plaques or intracellular inclusions [113] [8].
The strategic targeting of these aggregates, particularly the soluble oligomeric forms now recognized as the primary mediators of neurotoxicity, represents a promising therapeutic avenue. Monoclonal antibodies (mAbs) offer exceptional potential in this domain due to their high specificity, ability to be engineered for enhanced blood-brain barrier (BBB) penetration, and capacity to selectively neutralize toxic species while promoting clearance [114] [115]. This technical review comprehensively examines current antibody-based immunotherapies targeting protein aggregates, detailing their mechanisms, experimental methodologies, and applications within the broader context of proteostasis failure in neurodegenerative diseases.
The Aggregation Pathway and Molecular Targets
Structural Progression of Protein Aggregation
Protein aggregation follows a multi-stage pathway that begins with the partial unfolding of native monomers, leading to the exposure of aggregation-prone regions (APRs) and subsequent molecular assembly [113]. The process evolves through distinct intermediates:
- Partial Unfolding: Native proteins undergo conformational destabilization under stress conditions, exposing hydrophobic residues and β-strand motifs that are normally buried [113].
- Oligomerization: Misfolded monomers associate into soluble oligomeric species characterized by amorphous structures rich in exposed hydrophobic regions, rendering them highly reactive and toxic [8].
- Fibrillization: Oligomers undergo structural rearrangement into protofibrils and mature fibrils with extensive β-sheet content, eventually forming stable amyloid structures that can persist for decades in biological systems [8].
The aggregation process is governed by competing molecular-scale interactions that balance fold-favoring interactions (electrostatic attraction, hydrophobic interaction, hydrogen bonding) against unfolding-favoring interactions (geometric constraints, steric clash, electrostatic repulsion) [113].
Figure 1: The Protein Aggregation Pathway. The process begins with native monomer destabilization, progresses through toxic oligomer formation, and culminates in stable fibril and plaque deposition. Antibody therapeutic strategies target specific intermediates along this pathway.
Key Pathological Proteins in Neurodegenerative Diseases
Table 1: Major Aggregating Proteins in Neurodegenerative Diseases
| Protein Name | Primary Associated Disease(s) | Aggregate Morphology & Key Characteristics | Common Abbreviation |
|---|---|---|---|
| Amyloid-β | Alzheimer's disease (AD) | Forms extracellular senile plaques (SPs). Aβ42 isoform is more aggregation-prone and toxic than Aβ40. | Aβ |
| Tau | Alzheimer's Disease (AD), Pick's Disease, Progressive Supranuclear Palsy | Forms intracellular neurofibrillary tangles (NFTs). Hyperphosphorylated tau misfolds and aggregates into fibrils. | tau |
| α-synuclein | Parkinson's Disease (PD), Dementia with Lewy Bodies (DLB) | Forms neuronal Lewy Bodies in PD/DLB. Aggregation involves monomers → oligomers → protofibrils → mature fibrils. | α-syn, aSyn |
| Huntingtin | Huntington's Disease (HD) | Contains expanded polyglutamine (polyQ) tract leading to protein misfolding, aggregation, and intracellular inclusions. | HTT, Htt |
| TAR DNA-binding protein 43 | Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Lobar Degeneration | Normally nuclear, mis-localizes to cytoplasm in disease states where it aggregates and forms inclusions. | TDP-43 |
| PrPRes | Prion diseases | Forms protease-resistant aggregates that template further misfolding of native prion protein. | PrPSc |
Antibody Mechanisms in Aggregate Clearance
Molecular Mechanisms of Action
Therapeutic antibodies targeting protein aggregates employ multiple mechanistic strategies to counteract pathology, with different antibody species preferentially engaging specific mechanisms:
- Direct Neutralization: High-affinity binding to toxic oligomeric species neutralizes their biological activity by preventing interaction with cellular membranes and receptors, thereby abrogating synaptic toxicity [8] [115].
- Peripheral Sink Hypothesis: Antibodies sequester target proteins in the peripheral circulation, creating a concentration gradient that promotes efflux of aggregates from the central nervous system [114].
- Fc-Mediated Clearance: Antibodies opsonize pathological aggregates for phagocytosis by microglia via Fc receptor engagement, facilitating bulk clearance of amyloid plaques and other insoluble deposits [114].
- Sequestration and Disaggregation: Certain antibodies bind to fibrillar structures and accelerate their disassembly into less toxic, soluble forms that can be more readily cleared from the brain parenchyma [116].
- Prevention of Cell-to-Cell Transmission: Antibodies intercept prion-like propagation by binding to extracellular aggregates, preventing their uptake by healthy neurons and subsequent templated misfolding of endogenous proteins [8].
Blood-Brain Barrier Penetration Strategies
The blood-brain barrier represents a significant challenge for antibody therapeutics, with several engineering approaches employed to enhance CNS delivery:
- Receptor-Mediated Transcytosis: Bispecific antibodies like AK152 simultaneously target Aβ and BBB-expressed receptors, leveraging endogenous transport mechanisms to significantly enhance brain penetration [115].
- Fc Engineering: Modifications to the Fc region can enhance binding to transcytosis receptors while reducing effector functions that might contribute to inflammatory side effects [117].
- Affinity Optimization: Carefully tuned binding affinity balances target engagement with the ability to distribute beyond vascular compartments into brain parenchyma [117].
Figure 2: Antibody Mechanisms for Aggregate Clearance. Therapeutic antibodies employ multiple strategies including enhanced BBB penetration, direct target neutralization, Fc-mediated phagocytosis, and peripheral sink effects to combat protein aggregation pathology.
Clinically Approved and Investigational Antibodies
Anti-Aβ Antibodies for Alzheimer's Disease
Table 2: Clinically Approved Anti-Aβ Antibodies for Alzheimer's Disease
| Antibody Name | Target Epitope | Mechanism of Action | Clinical Efficacy | Safety Profile |
|---|---|---|---|---|
| Aducanumab | Aβ aggregates (particularly fibrils) | Selective targeting of aggregated forms, promotes Fc-mediated clearance | Mixed results across trials, some showing plaque reduction and slowing of clinical decline | ARIA-E: ~35% in APOE ε4 carriers, ~19% non-carriers; led to withdrawal |
| Lecanemab | Aβ protofibrils and soluble oligomers | Preferentially binds toxic protofibrils, neutralizes oligomeric toxicity | 27% slowing of clinical decline on CDR-SB over 18 months; robust plaque reduction | ARIA-E: ~21% in APOE ε4 carriers, ~10% in non-carriers |
| Donanemab | Aβ plaque (pyroglutamate form) | Targets a modified form of Aβ present in established plaques | 29-35% slowing of decline on iADRS; rapid plaque clearance | ARIA-E: ~27% in APOE ε4 carriers, ~13% in non-carriers |
| AK152 (bispecific) | Aβ oligomers + BBB receptor | Enhances brain penetration via receptor-mediated transcytosis | Preclinical models show accelerated plaque clearance and pathology reversal | Favorable preclinical profile; clinical trials ongoing |
Emerging Targets and Approaches
Beyond Aβ-directed therapies, numerous investigative approaches target other aggregating proteins:
- Anti-tau Antibodies: Target pathological forms of tau including phosphorylated species and oligomeric assemblies, aiming to prevent neurofibrillary tangle formation and cell-to-cell transmission [8] [112].
- Conformation-Selective Antibodies: Engineered to recognize β-sheet secondary structures common to many amyloidogenic proteins, enabling targeting of multiple pathological aggregates with a single therapeutic agent [112].
- α-Synuclein Targeting: Antibodies designed to intercept α-synuclein aggregation early in the pathway, preventing Lewy body formation and neuronal dysfunction in synucleinopathies [8].
Experimental Protocols and Methodologies
Production of Conformation-Specific Monoclonal Antibodies
The generation of antibodies selectively targeting pathological protein conformers requires specialized immunization and screening strategies:
Immunogen Preparation (p13Bri Method):
- Peptide Selection: Utilize a 13-amino acid peptide from the carboxyl terminus of British amyloidosis (ABri) with no homology to mammalian proteins to avoid autoimmune responses [112].
- Polymerization: Incubate peptide with 0.2% glutaraldehyde in 0.1M sodium carbonate buffer (pH 9.6) at 37°C with vigorous shaking (300 rpm) for 24 hours to form covalently-bound oligomers [112].
- Size Fractionation: Separate reaction products via size exclusion chromatography, collecting fractions between 10-100 kDa corresponding to soluble, stable oligomers with high β-sheet content [112].
- Structural Validation: Confirm β-sheet secondary structure using circular dichroism spectroscopy with characteristic minimum at 218 nm and maximum at 195 nm [112].
Hybridoma Generation and Screening:
- Immunization: Administer p13Bri immunogen (50μg/dose) to CD-1 mice via subcutaneous injection with Sigma Adjuvant at 2-week intervals for 12 weeks [112].
- Fusion Protocol: Fuse splenocytes from immunized mice with SP2/0-IL6 partner cells at 5:1 ratio using 50% PEG 1500, plate in HAT selection medium with 20% FBS [112].
- Conformational Screening: Screen hybridoma supernatants against multiple pathological oligomeric conformers (Aβ, tau, α-synuclein) using ELISA to identify clones with broad β-sheet specificity [112].
- Functional Characterization: Validate antibody specificity through immunohistochemistry on human AD brain sections, immunoblotting of oligomeric species, and neutralization assays in primary neuronal cultures [112].
In Vitro Aggregation and Antibody Intervention Assays
Quantitative assessment of antibody effects on aggregation kinetics employs standardized biochemical approaches:
Aβ Aggregation Kinetics Assay:
- Monomer Preparation: Dissolve recombinant Aβ42 in 10mM NaOH (1mg/mL), sonicate for 1 minute, then dilute in bicarbonate buffer (pH 9.6) to working concentrations (1.1-5μM) [116].
- Thioflavin T Monitoring: Add 20μM Thioflavin T (ThT) to Aβ solutions, aliquot 100μL/well in black 96-well plates with clear bottoms, and cover with sealing tape [116].
- Kinetic Measurements: Monitor ThT fluorescence (excitation 440nm, emission 485nm) every 10 minutes using a plate reader maintained at 37°C with continuous shaking between readings [116].
- Antibody Intervention: Pre-incubate therapeutic antibodies (0.1-10μM) with Aβ monomers for 1 hour prior to initiation of aggregation, or add at specified timepoints to assess stage-specific effects [116].
- Data Analysis: Fit fluorescence trajectories to sigmoidal curves to derive lag time, growth rate, and maximum fluorescence parameters for quantitative comparison between treatment conditions [116].
Mathematical Modeling of Aggregation Dynamics: Recent approaches employ systems biology frameworks to quantitatively model Aβ aggregation and antibody effects using ordinary differential equations that capture transitions between monomeric (M), oligomeric (O), protofibril (P), and fibrillar (F) states [116]:
[ \begin{aligned} \frac{dM}{dt} &= -k1M - k2MO - k3MF \ \frac{dO}{dt} &= k1M - k2MO - k4O - k7O + k5P \ \frac{dP}{dt} &= k4O - k5P - k6P + k8F \ \frac{dF}{dt} &= k6P - k8F - k_9[A]F \end{aligned} ]
Where [A] represents antibody concentration and rate constants (k1)-(k9) describe specific aggregation and clearance processes that can be estimated from experimental data [116].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Antibody-Based Aggregate Clearance Studies
| Reagent Category | Specific Examples | Function and Application | Technical Considerations |
|---|---|---|---|
| Aggregation-Prone Peptides/Proteins | Recombinant Aβ42, α-synuclein, tau | Substrates for in vitro aggregation studies; targets for antibody screening | Require careful handling to control pre-aggregation state; validate lot-to-lot consistency |
| Conformational Dyes | Thioflavin T (ThT), Congo Red | Detect β-sheet-rich aggregates via fluorescence or absorbance | ThT fluorescence increases with fibril mass but does not detect oligomers |
| Conformation-Specific Antibodies | A11 (oligomers), OC (fibrils) | Reference standards for specific aggregate morphologies | Lot variability can affect performance; require validation with appropriate controls |
| Cell Lines for Phagocytosis Assays | BV-2 microglia, HMC3 human microglia | Model Fc-mediated clearance mechanisms | Primary microglia better represent physiological responses but have limited expansion capacity |
| Blood-Brain Barrier Models | hCMEC/D3 cells, in vitro BBB co-cultures | Assess antibody penetration potential | Transwell systems with TEER measurement provide functional readout of barrier integrity |
| Animal Models of Aggregation | APP/PS1 mice (Aβ), P301S tau mice, Thy1-αSyn mice (Parkinson's) | In vivo evaluation of antibody efficacy and safety | Species differences in target sequence may affect antibody binding; immunodeficient models for human antibody testing |
| Analytical SEC Columns | Superdex 75, Superose 6 | Separate and characterize oligomeric species | Run at 4°C to minimize aggregation during separation; calibrate with appropriate standards |
Challenges and Future Directions
Current Limitations in Antibody-Based Immunotherapy
Despite promising clinical advances, significant challenges remain in the development of antibodies for aggregate clearance:
- Blood-Brain Barrier Penetration: Only 0.1-0.2% of systemically administered antibodies typically cross the intact BBB, necessitating high doses that increase cost and potential peripheral side effects [115].
- Target Selection Complexity: The precise pathological species responsible for neurodegeneration remains debated, with evidence supporting roles for both oligomers and fibrils in disease progression [8].
- Safety Considerations: Amyloid-Related Imaging Abnormalities (ARIA) represent a class-specific adverse effect of anti-Aβ antibodies, occurring more frequently in APOE ε4 carriers and requiring careful patient monitoring [114].
- Immunogenicity: Despite humanization strategies, anti-drug antibodies (ADAs) can develop against therapeutic mAbs, potentially altering pharmacokinetics, reducing efficacy, or causing hypersensitivity reactions [117].
Emerging Solutions and Innovative Approaches
Next-generation strategies aim to overcome current limitations through advanced engineering and novel target selection:
- Bispecific and Multispecific Platforms: Molecules like AK152 that simultaneously target pathological aggregates and BBB transporters demonstrate enhanced brain delivery and improved efficacy in preclinical models [115].
- Effector Function Engineering: Fc domain modifications to optimize phagocytic clearance while minimizing inflammatory activation and ARIA risk [117].
- Conformational Selectivity Expansion: Antibodies targeting generic amyloid motifs (β-sheet structures) rather than specific protein sequences offer potential for broader application across multiple proteinopathies [112].
- Combination Therapies: Strategic pairing of aggregate-targeting antibodies with enhancers of proteostasis network function (chaperones, autophagy activators) to address multiple nodes in the proteostasis network simultaneously [66].
The continued evolution of antibody-based therapeutics for protein aggregation disorders reflects an increasingly sophisticated understanding of both disease mechanisms and antibody engineering principles. As target engagement strategies advance and delivery challenges are addressed, immunotherapeutic approaches promise to play an expanding role in the treatment of neurodegenerative diseases characterized by protein misfolding and aggregation.
Cellular protein homeostasis, or proteostasis, encompasses the integrated biological pathways within a cell that regulate protein synthesis, folding, trafficking, and degradation [10]. In neurodegenerative diseases, this delicate balance is disrupted, leading to the accumulation of misfolded and aggregated proteins that are central to conditions such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) [10] [11]. The progressive decline of proteostasis network efficiency, particularly with aging or under pathological mutations, results in "proteostatic collapse," a state characterized by widespread accumulation of ubiquitinated protein inclusions and neuronal dysfunction [10]. This collapse represents a primary therapeutic target, and gene therapy has emerged as a clinically transformable approach to intervene in this process, leveraging advanced gene editing and delivery systems to address the molecular underpinnings of aging and neurodegeneration [118].
The significance of targeting proteostasis extends across multiple neurodegenerative proteinopathies, which share common mechanisms of pathology despite involving different aggregating proteins. Protein misfolding and aggregation initiate cascades of cellular toxicity, including synaptic dysfunction, mitochondrial impairment, activation of inflammatory responses, and ultimately neuronal death [11] [12]. A hallmark of these diseases is the prion-like propagation of misfolded proteins, where pathological protein species recruit their normally folded counterparts into toxic aggregates, enabling disease progression through connected brain regions [10] [119]. This review examines the evolving landscape of gene therapy strategies designed to reinforce the proteostasis network, with a focus on mechanistic approaches, experimental validation, and translational potential for treating neurodegenerative diseases.
Molecular Basis of Proteostatic Imbalance
Key Proteins and Aggregation Pathways
Neurodegenerative diseases are characterized by the aberrant misfolding and accumulation of specific proteins, each associated with distinct pathological signatures. Amyloid-β (Aβ) and tau proteins aggregate to form senile plaques and neurofibrillary tangles in Alzheimer's disease, while α-synuclein forms Lewy bodies in Parkinson's disease and related synucleinopathies [10] [120] [11]. In Huntington's disease, mutant huntingtin protein with expanded polyglutamine tracts forms intracellular aggregates, and in ALS, proteins such as TDP-43 and FUS accumulate in cytoplasmic inclusions [10] [12]. These aggregates share common structural features, particularly the cross-β-sheet architecture of amyloid fibrils, despite differences in their primary amino acid sequences [10].
The process of protein aggregation follows a predictable molecular pathway. Initially, native proteins undergo conformational changes that expose hydrophobic regions, leading to the formation of soluble oligomers. These oligomeric species are now widely considered to be the most neurotoxic forms, capable of disrupting synaptic function and cellular integrity [16]. Through further aggregation, these oligomers assemble into protofibrils and mature fibrils, which eventually deposit into larger insoluble aggregates [11] [12]. Recent research has revealed that the transition between these states is influenced by numerous cellular factors, including the concentration of metal ions, lipid membrane interactions, post-translational modifications, and the unique molecular environment surrounding the proteins [120] [119].
Cellular Clearance Mechanisms and Their Decline
The mammalian proteostasis network comprises several complementary systems for maintaining protein integrity, primarily through molecular chaperones and protein degradation pathways. Molecular chaperones, including heat shock proteins (HSPs) and chaperones linked to protein synthesis (CLIPs), oversee protein quality control by facilitating proper folding, preventing aggregation, and targeting irreversibly damaged proteins for degradation [10]. Two major degradation systems operate in parallel: the ubiquitin-proteasome system (UPS) for short-lived and soluble proteins, and the autophagy-lysosome pathway for larger protein complexes and organelles [10] [11]. Chaperone-mediated autophagy (CMA) represents a selective degradation pathway that directly shuttles specific protein substrates across the lysosomal membrane [11].
During aging and in neurodegenerative conditions, these essential clearance mechanisms become progressively impaired. The age-related decline of proteostasis network components reduces cellular capacity to handle misfolded proteins, creating a vulnerable environment for aggregation [118] [10]. Notably, in the aging human brain, a subset of chaperones (primarily CLIPs) becomes repressed, while stress-responsive chaperones are induced, mimicking a state of chronic proteotoxic stress [10]. This impairment is further exacerbated by the direct inhibitory effects of protein aggregates on proteasomal function and autophagic flux, establishing a vicious cycle of accumulating proteotoxicity [10] [11]. Additionally, disease-associated mutations in proteins such as α-synuclein can directly impair CMA, while aggregates can physically obstruct the UPS, thereby accelerating pathological progression [11].
Table 1: Major Protein Aggregates in Neurodegenerative Diseases
| Disease | Aggregating Protein(s) | Pathological Structure |
|---|---|---|
| Alzheimer's disease | Amyloid-β (Aβ), Tau | Senile plaques, Neurofibrillary tangles |
| Parkinson's disease | α-Synuclein | Lewy bodies |
| Huntington's disease | Huntingtin (with expanded polyglutamine) | Nuclear and cytoplasmic inclusions |
| Amyotrophic lateral sclerosis (ALS) | TDP-43, SOD1, FUS | Cytoplasmic inclusions |
| Prion diseases | Prion protein (PrPSc) | Amyloid plaques, Spongiform degeneration |
Gene Therapy Strategies for Proteostatic Correction
Enhancing Protein Clearance Pathways
Gene therapy approaches aimed at bolstering the cellular capacity to clear misfolded proteins represent a promising strategic direction. Augmenting autophagy through overexpression of key regulators has demonstrated efficacy in multiple disease models. Strategies include delivering genes encoding for autophagy initiators such as transcription factor EB (TFEB), which activates lysosomal biogenesis and enhances the clearance of aggregated proteins including α-synuclein and mutant huntingtin [16] [11]. Similarly, upregulation of the Keap1-Nrf2-ARE pathway via gene therapy approaches enhances the expression of antioxidant and cytoprotective genes, indirectly supporting proteostasis by reducing oxidative damage to the proteostasis machinery [11].
The ubiquitin-proteasome system can also be targeted through gene therapeutic interventions. Overexpression of specific E3 ubiquitin ligases can facilitate the targeting of disease proteins for degradation, while engineering proteasome subunits with enhanced activity may improve clearance capacity [16]. Another innovative approach involves modulating chaperone networks through the delivery of molecular chaperones such as Hsp70, Hsp40, or small heat shock proteins that can selectively interact with misfolded disease proteins and facilitate their refolding or degradation [10] [11]. These approaches are particularly promising given the documented repression of certain chaperone networks in the aging brain and in neurodegenerative conditions [10].
Targeting Aggregation-Prone Proteins Directly
Direct targeting of disease-associated proteins represents another major gene therapy strategy. RNA interference (RNAi) approaches utilize short hairpin RNAs (shRNAs) or small interfering RNAs (siRNAs) to selectively silence the expression of genes encoding aggregation-prone proteins. This strategy has shown promise in reducing the production of mutant huntingtin in Huntington's disease models, α-synuclein in Parkinson's disease models, and tau in models of tauopathies [16] [11]. Antisense oligonucleotides (ASOs) represent another modality for gene suppression, with several candidates advancing to clinical trials for diseases including ALS and Huntington's disease [16].
For monogenic neurodegenerative diseases caused by specific mutations, gene editing technologies such as CRISPR-Cas9 offer the potential for permanent correction. This approach can be used to directly disrupt mutant genes or to correct disease-causing mutations at the genomic level [118]. Alternatively, gene replacement strategies can be employed to deliver wild-type copies of genes to compensate for loss-of-function mutations, as has been explored for certain forms of Parkinson's disease linked to GBA1 mutations [118] [16]. These direct targeting approaches are complemented by immunotherapy-based gene therapy, which involves the delivery of gene constructs encoding antibody fragments (intrabodies) that selectively bind to and neutralize toxic conformations of misfolded proteins, thereby preventing their aggregation and cellular toxicity [16].
Restoring Metabolic and Immune Homeostasis
Beyond direct protein targeting, gene therapy strategies are being developed to address the broader cellular dysfunctions associated with proteostatic collapse. Metabolic reprogramming approaches aim to restore energy homeostasis in stressed neurons, as metabolic impairment is both a cause and consequence of proteostatic failure [118]. This includes enhancing mitochondrial function through delivery of genes involved in mitochondrial biogenesis and quality control, such as PGC-1α, which has shown neuroprotective effects in models of Parkinson's and Alzheimer's diseases [118].
Similarly, modulating neuroimmune responses represents a promising avenue for intervention. Chronic neuroinflammation driven by activated microglia and astrocytes significantly contributes to proteostatic disruption in neurodegenerative diseases [118] [11]. Gene therapy approaches to deliver anti-inflammatory cytokines such as IL-10 or to express immunomodulatory factors can shift the brain environment toward a more homeostatic state, indirectly supporting protein quality control mechanisms [118]. The interplay between proteostasis and these broader physiological processes highlights the potential of multi-targeted gene therapy approaches that address both specific protein pathologies and the overall cellular environment conducive to neurodegeneration.
Table 2: Gene Therapy Approaches for Proteostatic Correction
| Strategy | Molecular Targets | Potential Applications |
|---|---|---|
| Enhanced Autophagy | TFEB, ATG genes, p62 | PD, AD, HD |
| Chaperone Network Enhancement | Hsp70, Hsp40, Hsp90 | PD, AD, PolyQ diseases |
| RNA Interference | Mutant HTT, SNCA, MAPT | HD, PD, Tauopathies |
| Gene Editing | Disease-causing mutations via CRISPR-Cas9 | Monogenic forms of ND |
| Immunotherapy | Aβ, α-synuclein, tau via intrabodies | AD, PD, Tauopathies |
| Metabolic Reprogramming | PGC-1α, SIRT1, mitochondrial genes | PD, AD, ALS |
Experimental Models and Methodologies
In Vitro Aggregation and Toxicity Assays
The investigation of proteostatic mechanisms and the validation of therapeutic interventions rely heavily on robust experimental models. In vitro aggregation assays utilizing recombinant proteins provide controlled systems for studying the fundamental processes of protein misfolding. The Thioflavin T (ThT) fluorescence assay is widely employed to monitor amyloid fibril formation in real-time, as ThT exhibits enhanced fluorescence upon binding to cross-β-sheet structures [12]. Similarly, turbidity measurements and static light scattering can quantify the formation of large aggregates, while atomic force microscopy (AFM) and electron microscopy offer direct visualization of oligomeric and fibrillar species [119] [12].
To assess the functional consequences of protein aggregation and the efficacy of interventions, researchers employ various cell-based toxicity assays. Primary neuronal cultures or neuroblastoma cell lines transfected to express disease-associated proteins are used to model intracellular aggregation. Key endpoints include cell viability measurements (MTT, MTS, or Alamar Blue assays), apoptotic markers (caspase activation, TUNEL staining), mitochondrial function (JC-1 or TMRE staining for membrane potential), and oxidative stress (DCFDA for reactive oxygen species) [12]. High-content screening platforms combining automated microscopy with multi-parameter analysis enable comprehensive assessment of morphological and functional changes in response to proteostatic challenges [12].
In Vivo Modeling and Therapeutic Validation
Animal models are essential for evaluating gene therapy strategies in complex biological systems. Transgenic mouse models overexpressing human disease genes with pathogenic mutations (e.g., APP/PS1 for AD, A53T α-synuclein for PD, R6/2 for HD) recapitulate key aspects of protein aggregation and neurodegeneration [10]. These models enable the assessment of therapeutic interventions on behavioral outcomes (cognitive and motor function), neuropathology (aggregate burden, neuronal loss), and biochemical markers [10]. More recently, knock-in models with endogenous expression of mutant proteins provide more physiologically relevant systems for therapeutic development [16].
The evaluation of gene therapy efficacy in these models involves stereotactic intracranial delivery of viral vectors (typically adeno-associated viruses or lentiviruses) carrying therapeutic transgenes to affected brain regions. Critical analytical methods include immunohistochemical quantification of protein aggregates, western blot analysis of protein expression and modification, RNA sequencing to assess transcriptional responses, and behavioral testing to determine functional recovery [16] [119]. Longitudinal monitoring using live-animal imaging techniques such as magnetic resonance imaging (MRI) and positron emission tomography (PET) with aggregate-specific tracers enables non-invasive assessment of disease progression and therapeutic effects [16].
Research Reagent Solutions
Table 3: Essential Research Reagents for Proteostasis and Gene Therapy Studies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Aggregation Detection Dyes | Thioflavin T, Thioflavin S, Congo Red | Amyloid fibril quantification in solution and tissue |
| Protein-Specific Antibodies | Anti-phospho-tau, Anti-oligomeric Aβ, Anti-ubiquitin | Immunodetection of pathological protein species |
| Viral Vector Systems | AAV1/2/5/8/9, Lentivirus, Adenovirus | In vitro and in vivo gene delivery |
| Gene Editing Tools | CRISPR-Cas9 plasmids, gRNA libraries, ZFNs, TALENs | Targeted genome modification |
| Proteostasis Reporters | GFPu, Hsp70 reporters, FRET-based proteostasis sensors | Monitoring protein folding environment and degradation capacity |
| Cell Lines | SH-SY5Y, PC12, HEK293, Primary neuronal cultures | In vitro modeling of protein aggregation and toxicity |
Visualization of Key Pathways and Workflows
Proteostasis Network and Therapeutic Intervention Points
Diagram 1: Proteostasis Network with Gene Therapy Intervention Points. This diagram illustrates the cellular protein quality control system and key points for therapeutic intervention using gene therapy approaches to enhance component functions.
Experimental Workflow for Gene Therapy Validation
Diagram 2: Gene Therapy Validation Workflow. This diagram outlines the sequential process for developing and validating gene therapy approaches for proteostatic correction, from target identification through clinical translation.
Gene therapy represents a paradigm-shifting approach for addressing the fundamental proteostatic imbalances underlying neurodegenerative diseases. Current strategies encompass a diverse range of interventions, from enhancing protein clearance mechanisms to directly targeting aggregation-prone proteins and restoring broader cellular homeostasis [118] [16] [11]. The continued refinement of vector systems for efficient and targeted delivery, combined with increasingly precise gene editing technologies, promises to overcome current limitations in therapeutic specificity and durability. Furthermore, the development of sensitive biomarkers for patient stratification and treatment monitoring will be essential for translating these approaches to clinical practice [16].
Looking ahead, the field is moving toward multi-targeted combination therapies that address proteostatic collapse at multiple levels simultaneously [11]. The integration of gene therapies with small molecule proteostasis regulators represents a particularly promising direction. Additionally, advances in our understanding of the structural biology of protein aggregates, facilitated by technologies such as cryo-electron microscopy, are revealing new therapeutic targets for intervention [119]. As gene therapy technologies mature and our comprehension of proteostatic networks deepens, these innovative approaches hold exceptional promise for developing effective treatments for neurodegenerative diseases that have thus far remained intractable to conventional therapeutic strategies.
The accumulation of misfolded and aggregated proteins is a hallmark of numerous neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and Huntington's disease. Cellular defense mechanisms against proteotoxicity center on molecular chaperones, particularly heat shock proteins (HSPs), which prevent protein aggregation and promote refolding or degradation of damaged proteins. This whitepaper provides an in-depth technical analysis of pharmacological strategies to induce the heat shock response by activating Heat Shock Factor 1 (HSF1), the master regulator of chaperone expression. We present quantitative comparisons of HSF1 activators, detailed experimental protocols for assessing induction efficacy, and visualization of the underlying molecular mechanisms. For research purposes, targeting molecular chaperone induction represents a promising therapeutic strategy to restore proteostasis in neurodegenerative conditions.
Molecular chaperones, predominantly heat shock proteins (HSPs), constitute a highly conserved cellular defense system that maintains protein homeostasis (proteostasis) by facilitating the correct folding of nascent polypeptides, refolding of misfolded proteins, and directing irreversibly damaged proteins for degradation [31] [121]. Under physiological conditions, chaperones are integral to cellular viability; under stress conditions, their expression is significantly upregulated to counter proteotoxic damage [122]. The accumulation of misfolded and aggregated proteins is a defining feature of neurodegenerative diseases, where specific proteins such as amyloid-β and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, and huntingtin in Huntington's disease form toxic species that disrupt neuronal function and viability [31]. In many neurodegenerative contexts, the expression or function of protective chaperones is compromised, creating a vicious cycle of accumulating proteotoxicity [122] [31]. Consequently, the targeted induction of molecular chaperones presents a compelling strategy to bolster the cellular defense system, potentially slowing or preventing the progression of these devastating disorders.
The Heat Shock Response Pathway: Mechanism of Chaperone Induction
The transcriptional activation of HSP genes is primarily regulated by Heat Shock Factor 1 (HSF1). Under normal conditions, HSF1 exists as an inactive monomer in the cytoplasm and nucleus, bound by a repressive complex that includes HSP70 and HSP90 [122]. Upon proteotoxic stress—such as heat shock, oxidative stress, or the presence of misfolded proteins—these chaperones are recruited to misfolded clients, thereby releasing HSF1. This allows HSF1 to trimerize, undergo post-translational modifications (including phosphorylation and acetylation), and accumulate in the nucleus. There, it binds to specific DNA sequences known as Heat Shock Elements (HSEs) in the promoters of target genes, driving the expression of various HSPs [122] [123]. The newly synthesized HSPs then function to restore proteostasis and, through a negative feedback loop, bind to HSF1 again to attenuate the response [122].
The following diagram illustrates this central regulatory pathway:
Quantitative Comparison of HSF1 Activators
A systematic quantitative comparison of published HSF1 activators is essential for selecting appropriate compounds for research. Using a stable HSF-responsive dual-luciferase reporter cell line (e.g., HEK293 cells with a 12xHSE-NanoLuc construct), researchers can directly compare induction potency and specificity [123]. The table below summarizes key quantitative data for a range of activators, highlighting their mechanisms and therapeutic potential.
Table 1: Quantitative Comparison of HSF1 Activators in a Luciferase Reporter Assay
| Activator | Proposed Mechanism | Relative HSF1 Activation (Fold vs. Control) | Therapeutic Index (EC₅₀ vs. IC₅₀ Viability) | Key Findings and Notes |
|---|---|---|---|---|
| Geldanamycin | HSP90 inhibitor [123] | >50-fold [123] | Low [123] | Potent but high toxicity; derivatives in development. |
| SNX-2112 | HSP90 inhibitor [123] | High (specific data not shown) | Moderate [123] | Prodrug SNX-5422 tested in clinical trials for cancer. |
| Arimoclomol | HSF1 co-inducer [123] | ~5-10 fold [123] | High [123] | Amplifies HSR during stress; promising in clinical trials for neurodegenerative diseases (e.g., ALS, IBM). |
| BGP-15 | HSF1 co-inducer [123] | Moderate [123] | High [123] | Well-tolerated; can enhance stress-induced chaperone expression. |
| Cadmium Sulfate | Heavy metal stressor [123] | >20-fold [123] | Very Low [123] | Powerful inducer but severely cytotoxic. |
| Mefenamic Acid | NSAID/HSF1 activator [123] | ~4-5 fold [123] | Moderate [123] | Repurposed drug with moderate induction capacity. |
| Sulindac | NSAID/HSF1 activator [123] | ~3-4 fold [123] | Moderate [123] | Repurposed drug with moderate induction capacity. |
| VER-155008 | HSP70 inhibitor [123] | Moderate [123] | Low [123] | Induces HSR by inhibiting HSP70 feedback; toxicity concerns. |
Key Insights from Comparative Data
- Mechanistic Classes: Activators fall into two broad categories: direct stressors (e.g., cadmium, H₂O₂) and pharmacologic modulators. The latter includes HSP90 inhibitors (e.g., Geldanamycin), which disrupt the repressive complex on HSF1, and co-inducers (e.g., Arimoclomol), which enhance the HSR during pre-existing stress [123].
- Potency vs. Specificity: A critical finding is the inverse correlation often observed between the potency of HSF1 activation and compound specificity. Powerful inducers like geldanamycin and cadmium sulfate frequently exhibit significant cytotoxicity, limiting their therapeutic utility. In contrast, co-inducers like arimoclomol and BGP-15 show more favorable toxicity profiles, making them more suitable candidates for chronic diseases [123].
- Tissue-Specific Effects: The data also indicate that some activators may exhibit tissue-specific differences in their efficacy, underscoring the importance of validating results in disease-relevant cell models [123].
Experimental Protocol: Quantifying HSF1 Activation
To ensure reproducible assessment of potential HSF1 activators, the following detailed protocol is provided.
HSF-Responsive Luciferase Reporter Assay
Objective: To quantitatively measure the activation of the Heat Shock Response pathway by test compounds.
Materials:
- Reporter Cell Line: Stably transfected HEK293 or HeLa cells containing a plasmid with multiple Heat Shock Elements (HSEs) driving the expression of a luciferase reporter (e.g., NanoLuc) [123].
- Test Compounds: A selection of activators from Table 1 (e.g., geldanamycin, arimoclomol). Prepare stock solutions in recommended solvents (e.g., DMSO, water) and store at -20°C.
- Controls: Vehicle control (e.g., DMSO), positive control (e.g., 42°C heat shock for 1 hour, or 100 µM cadmium sulfate).
- Equipment: Cell culture incubator (37°C, 5% CO₂), luminescence plate reader, cell viability assay kit (e.g., MTT, CellTiter-Glo).
Procedure:
- Cell Seeding: Seed reporter cells into 96-well plates at a density that will yield ~80% confluence at the time of assay (e.g., 20,000 cells/well for HEK293). Culture for 48 hours.
- Compound Treatment: Prepare serial dilutions of test compounds in complete culture medium. Replace the medium in the assay plates with the compound-containing medium. Incubate the cells for a standardized period, typically 24 hours [123].
- Luciferase Measurement: Following incubation, lyse the cells according to the instructions of the dual-luciferase assay system. Transfer the lysate to a white-walled plate and measure the luminescence signal using a plate reader.
- Viability Assessment: In parallel, perform a cell viability assay (e.g., CellTiter-Glo) on treated cells to normalize luminescence data to cell number and calculate a therapeutic index (EC₅₀ for induction vs. IC₅₀ for viability) [123].
- Data Analysis: Normalize luminescence of treated samples to the vehicle control. Plot dose-response curves to determine the half-maximal effective concentration (EC₅₀) for HSF1 activation for each compound.
The workflow for this protocol is summarized below:
Validation by Endogenous HSP Expression
Objective: To confirm that HSF1 activation, as measured by the reporter, leads to increased expression of endogenous heat shock proteins.
Procedure:
- Treat wild-type cells (e.g., HeLa, WI38 fibroblasts) with the top candidate compounds identified in the reporter screen.
- After 24 hours, harvest cell lysates and perform Western blotting using antibodies against major HSPs, notably HSP72 (HSPA1A)—the most strongly upregulated HSP—as well as HSP27 and HSP90 [123].
- Compare the protein levels to those in vehicle-treated and heat-shocked positive control cells.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful research in this field relies on a suite of essential reagents and tools.
Table 2: Essential Research Reagents for Studying HSP Induction
| Reagent / Tool | Function and Application | Example Use Case |
|---|---|---|
| HSF-Reporter Cell Line | Engineered cells (e.g., HEK293, HeLa) with HSE-driven luciferase for quantitative HSF1 activity screening. | Primary high-throughput screen for novel HSF1 activators [123]. |
| HSP90 Inhibitors (e.g., Geldanamycin, SNX-2112) | Tool compounds that disrupt the HSP90-HSF1 complex, leading to robust HSF1 activation. | Positive control in reporter assays; studying HSP90's role in HSF1 regulation [123]. |
| Co-Inducers (e.g., Arimoclomol, BGP-15) | Compounds that potentiate the HSR under stress conditions without being strong inducers alone. | Investigating therapeutic strategies for chronic neurodegenerative disease models [123]. |
| HSP-Specific Antibodies | Validate endogenous protein levels of key HSPs (e.g., HSP70, HSP27, HSP90) via Western blot. | Confirming transcriptional activation results at the protein level [123]. |
| Viability Assay Kits (e.g., MTT, CellTiter-Glo) | Assess cytotoxicity of potential inducers to calculate a therapeutic index. | Differentiating specific HSF1 activators from general cellular stressors [123]. |
The strategic induction of molecular chaperones by activating HSF1 represents a mechanistically rational approach to counter protein misfolding and aggregation in neurodegenerative diseases. Quantitative comparisons reveal a diverse pharmacological landscape, with compounds ranging from potent but toxic HSP90 inhibitors to safer, more specific co-inducers like arimoclomol. The experimental frameworks and tools detailed in this whitepaper provide a foundation for rigorous preclinical research. As the structural and mechanistic understanding of chaperone systems like Hsp70-Hsp40 deepens, new opportunities for targeted therapeutic intervention will continue to emerge [28] [124]. The ongoing clinical evaluation of chaperone-inducing compounds holds significant promise for developing the first disease-modifying treatments that target the root cause of proteotoxicity in neurodegeneration.
Comparative Efficacy of UPS Enhancement Versus Autophagy Activation
The accumulation of misfolded and aggregated proteins is a defining pathological feature of many neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD). Cellular protein quality control mechanisms, particularly the ubiquitin-proteasome system (UPS) and autophagy, represent critical therapeutic targets for clearing these toxic protein species. This review provides a comprehensive technical analysis comparing the therapeutic efficacy of enhancing UPS function versus activating autophagy pathways. We examine molecular mechanisms, experimental evidence, therapeutic strategies, and technical methodologies, presenting structured data and visualization tools to guide research and drug development efforts aimed at restoring proteostasis in neurodegenerative contexts.
Neurodegenerative diseases are characterized by the accumulation of misfolded proteins that form cytotoxic aggregates within neurons. In AD, extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein drive pathology [125]. PD involves the aggregation of α-synuclein into Lewy bodies, while HD is caused by polyglutamine (polyQ) expansions in the huntingtin protein that lead to nuclear inclusions [126]. Amyotrophic lateral sclerosis (ALS) is associated with aggregates of TAR DNA-binding protein 43 (TDP-43) or superoxide dismutase 1 (SOD1) [125] [126].
These disease-associated proteins share a tendency to misfold and form β-sheet-enriched oligomers that are partially resistant to cellular degradation systems [126]. Post-mitotic neurons are particularly vulnerable to proteostasis failure as they cannot dilute accumulated toxic proteins through cell division [126]. The unique architecture of neurons, with extensive dendrites and axons, further complicates protein quality control as aggregates must be transported long distances to the lysosome-rich cell body for degradation [125] [126]. Aging compounds these challenges through progressive decline in both UPS and autophagic activity [126].
The ubiquitin-proteasome system (UPS) and autophagy represent the two primary proteolytic pathways responsible for maintaining protein homeostasis. The UPS predominantly degrades short-lived soluble proteins, while autophagy eliminates long-lived proteins, insoluble aggregates, and damaged organelles [127] [126]. This review systematically compares therapeutic strategies targeting these two systems, evaluating their respective efficacy in combating proteinopathies.
Molecular Mechanisms and Comparative Functions
The Ubiquitin-Proteasome System (UPS)
The UPS is a sophisticated ATP-dependent proteolytic machinery responsible for the targeted degradation of short-lived proteins and soluble misfolded proteins [127] [128]. Degradation via UPS involves two key sequential steps: ubiquitination and proteasomal degradation [128] [129].
Ubiquitination Cascade: Ubiquitin modification involves a hierarchical enzyme system:
- E1 (ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent manner [128] [129]
- E2 (ubiquitin-conjugating enzyme): Accepts activated ubiquitin from E1 [128] [129]
- E3 (ubiquitin ligase): Recognizes specific substrates and facilitates ubiquitin transfer [128] [129]
The human genome encodes approximately 35 E2 enzymes and over 600 E3 ligases, providing exquisite substrate specificity [128]. E3 ligases, such as the HECT family members HUWE1 and KLHL8, have been implicated in neurodegenerative pathologies [130].
Proteasome Structure and Function: The 26S proteasome comprises a 20S core particle (CP) capped by 19S regulatory particles (RP) [128]. The 20S CP forms a barrel-like structure with outer α-rings controlling entry and inner β-rings housing proteolytic activities [127] [128]. The 19S RP recognizes ubiquitylated substrates, unfolds them, and translocates them into the proteolytic chamber [127] [128]. Proteasomal peptides are further processed into amino acids by cellular peptidases [127].
Ubiquitin Signaling: Ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) that enable formation of structurally and functionally distinct polyubiquitin chains [127] [131]. K48-linked chains represent the canonical signal for proteasomal degradation, while K63-linked chains typically mediate non-proteolytic functions, including autophagy targeting [127] [131].
Autophagy-Lysosome Pathway
Autophagy is a catabolic process that delivers cytoplasmic components to lysosomes for degradation. Three primary forms exist: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [125] [126]. Macroautophagy (hereafter autophagy) is responsible for degrading large protein aggregates and damaged organelles [127] [125].
Autophagy Initiation and Regulation: Autophagy induction converges on mTORC1, which integrates signals from nutrient availability, growth factors, and cellular energy status [127]. Under nutrient-replete conditions, mTORC1 phosphorylates and inhibits the ULK1/2 kinase complex. During starvation or stress, mTORC1 inhibition permits ULK1/2 activation, initiating autophagosome formation [127] [125]. A class III phosphatidylinositol 3-kinase (PI3K) complex containing VPS34 and Beclin1 generates phosphatidylinositol 3-phosphate (PtdIns3P), recruiting downstream autophagy components [127] [125].
Autophagosome Formation and Cargo Degradation: Autophagosome biogenesis involves two ubiquitin-like conjugation systems:
- ATG12-ATG5-ATG16L1 complex formation
- Microtubule-associated protein 1 light chain 3 (LC3) conversion to lipidated LC3-II [127] [125]
Mature autophagosomes fuse with lysosomes to form autolysosomes, where sequestered cargo is degraded by lysosomal hydrolases and components are recycled [127] [125].
Table 1: Key Characteristics of UPS and Autophagy
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagy |
|---|---|---|
| Primary Substrates | Short-lived proteins, soluble misfolded proteins [127] | Long-lived proteins, insoluble aggregates, damaged organelles [127] [126] |
| Degradation Capacity | Limited to unfolded polypeptides [126] | Large protein aggregates, whole organelles [127] [126] |
| Selectivity Mechanism | Ubiquitin tags (primarily K48-linked) [127] [131] | Ubiquitin tags (primarily K63-linked), receptor proteins (e.g., p62) [127] |
| Cellular Energy Requirements | ATP-dependent [129] | ATP-dependent [125] |
| Processing Output | Small peptides (3-25 amino acids) [127] | Amino acids, fatty acids, nucleotides [127] |
| Neuronal Vulnerability | High (particularly to aging) [126] | High (due to neuronal architecture) [125] [126] |
Therapeutic Strategies and Experimental Evidence
UPS Enhancement Approaches
UPS enhancement strategies face the challenge of activating a complex multi-enzyme system without disrupting its specificity. Several approaches have shown promise:
Proteasome Activators: Research has identified compounds that enhance proteasome activity, potentially compensating for age-related decline in proteolytic function. These activators work through various mechanisms, including stabilization of the proteasome complex and facilitation of substrate entry [128].
Ubiquitin Ligase Modulation: Targeting specific E3 ubiquitin ligases represents a precision medicine approach for neurodegenerative diseases. For instance, inhibiting HECT-E3 ligases with compound 056 (4-(((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-N-(pyridin-4-yl)benzamide) demonstrated neuroprotective effects in a traumatic optic neuropathy model, preserving retinal ganglion cells and visual function [130].
Transcription Factor Activation: Enhancing the expression of proteasome subunits through transcription factor activation (e.g., NRF1, NRF2) may boost proteasome biogenesis and activity, potentially increasing degradation capacity for misfolded proteins [129].
Autophagy Activation Strategies
Autophagy activation has emerged as a promising therapeutic strategy for clearing aggregated proteins in neurodegenerative diseases:
mTOR-Dependent Inducers: Rapamycin and its analogs (rapalogs) inhibit mTORC1, leading to ULK1/2 activation and autophagy induction. Rapamycin treatment has been shown to enhance autophagic clearance of protein aggregates in various models of neurodegeneration [125].
mTOR-Independent Inducers: Several compounds induce autophagy through mTOR-independent pathways, including:
- Trehalose: Disinhibits autophagy by activating TFEB
- Lithium: Reduces inositol levels, affecting calcium signaling
- Carbamazepine: Reduces inositol-1,4,5-trisphosphate (IP3) levels [125]
These compounds avoid potential side effects associated with mTOR inhibition while still promoting autophagic flux.
Late-Stage Autophagy Enhancement: In AD, accumulation of autophagic vacuoles results from impaired clearance rather than reduced formation. In such cases, enhancing later stages of autophagy (e.g., autophagosome-lysosome fusion) may be more beneficial than general autophagy induction [125].
Comparative Efficacy in Disease Models
Table 2: Therapeutic Efficacy of UPS Enhancement vs. Autophagy Activation in Neurodegenerative Disease Models
| Disease Model | UPS-Targeted Approach | Autophagy-Targeted Approach | Comparative Outcomes |
|---|---|---|---|
| Alzheimer's Disease | Limited efficacy due to proteasome inhibition by tau aggregates [126] | Rapamycin enhances Aβ and tau clearance; late-stage enhancement more effective [125] | Autophagy preferred due to capacity for aggregate clearance [125] [126] |
| Parkinson's Disease | Parkin gene therapy shows potential [131] | Rapamycin and mTOR-independent inducers enhance α-synuclein clearance [125] [126] | Context-dependent; autophagy for aggregate clearance, UPS for specific substrates [131] [126] |
| Huntington's Disease | Proteasome activators show limited efficacy against polyQ aggregates [126] | Strong evidence for autophagy inducers clearing mutant huntingtin [125] [126] | Autophagy preferred for large aggregates [126] |
| Traumatic Optic Neuropathy | HECT-E3 ligase inhibition with compound 056 promotes RGC survival [130] | Autophagy upregulation protects RGC soma after injury [130] | Combined approach most beneficial [130] |
| Multiple Myeloma | Proteasome inhibitors (ixazomib) standard care [132] | Autophagy inhibition enhances proteasome inhibitor efficacy [132] | Dual targeting synergistic [132] |
Experimental Protocols and Methodologies
Assessing UPS Function
Reporter-Based UPS Activity Assays:
- Principle: Fluorescent proteins (e.g., GFP, RFP) fused to degradation signals (e.g., CL1 degron) are constitutively expressed and degraded by UPS. UPS impairment increases fluorescence intensity.
- Protocol:
- Transfert cells with ubiquitin-proteasome reporter constructs (e.g., GFPu, RFPu)
- Treat with experimental compounds or conditions
- Measure fluorescence intensity via flow cytometry or fluorescence microscopy
- Normalize to protein concentration or control fluorescence
- Include proteasome inhibitor controls (e.g., MG132) to confirm UPS dependence
Proteasome Activity Assays:
- Principle: Fluorogenic substrates specific to different proteasome active sites (chymotrypsin-like, trypsin-like, caspase-like) measure proteasomal peptidase activity.
- Protocol:
- Prepare cell lysates in non-ionic detergent-containing buffer
- Incubate with fluorogenic substrates: Suc-LLVY-AMC (chymotrypsin-like), Z-ARR-AMC (trypsin-like), Z-LLE-AMC (caspase-like)
- Measure fluorescence emission over time (excitation ~380 nm, emission ~460 nm)
- Calculate activity relative to protein content and inhibitor controls
Immunoblotting for Polyubiquitinated Proteins:
- Principle: UPS impairment increases cellular levels of polyubiquitinated proteins.
- Protocol:
- Resolve proteins by SDS-PAGE under denaturing conditions
- Transfer to PVDF or nitrocellulose membranes
- Probe with anti-ubiquitin antibodies (e.g., FK1, FK2)
- Detect with HRP-conjugated secondary antibodies and chemiluminescence
- Normalize to loading controls (e.g., actin, tubulin)
Monitoring Autophagic Flux
LC3 Immunoblotting Turnover Assay:
- Principle: Autophagic flux is measured by comparing LC3-II levels with and without lysosomal inhibition.
- Protocol:
- Treat cells with experimental conditions with/without lysosomal inhibitors (bafilomycin A1 100 nM, chloroquine 50 μM)
- Prepare cell lysates in RIPA buffer
- Perform SDS-PAGE and immunoblot for LC3
- Quantify LC3-II bands normalized to loading controls
- Calculate autophagic flux as: LC3-II (+inhibitor) - LC3-II (-inhibitor)
GFP-LC3 Puncta Formation Assay:
- Principle: During autophagy, LC3 translocates to autophagosomal membranes, forming detectable puncta.
- Protocol:
- Seed cells expressing GFP-LC3 on glass coverslips
- Treat with experimental conditions with/without lysosomal inhibitors
- Fix with 4% paraformaldehyde, permeabilize with 0.1% Triton X-100
- Mount with anti-fade medium containing DAPI
- Visualize by fluorescence microscopy and quantify GFP-LC3 puncta per cell
Tandem Fluorescent LC3 (mRFP-GFP-LC3) Assay:
- Principle: GFP signal is quenched in acidic lysosomes while mRFP remains stable, differentiating autophagosomes (yellow) from autolysosomes (red).
- Protocol:
- Transfert cells with mRFP-GFP-LC3 construct
- Treat with experimental conditions
- Fix and image by confocal microscopy
- Quantify yellow (autophagosomes) and red (autolysosomes) puncta
- Calculate autophagic flux as red:yellow puncta ratio
Long-Lived Protein Degradation Assay:
- Principle: Measures bulk autophagic degradation of long-lived proteins.
- Protocol:
- Label proteins with [³H]-leucine for 24 hours
- Chase with excess unlabeled leucine for 4 hours to degrade short-lived proteins
- Treat with experimental conditions for 2-4 hours
- Measure TCA-soluble radioactivity in medium (degraded proteins)
- Express as percentage of total cellular radioactivity
Assessing Therapeutic Efficacy in Neurodegeneration Models
Neuronal Survival and Function Assays:
- Retinal Ganglion Cell (RGC) Survival in Optic Neuropathy Models:
- Perform retrograde labeling of RGCs with fluorogold
- Induce injury (e.g., optic nerve crush)
- Administer therapeutic compounds
- Quantify surviving RGCs per retina [130]
- Flash Visual Evoked Potentials (FVEPs):
- Implant recording electrodes in visual cortex
- Deliver light flashes of controlled intensity and duration
- Record cortical responses
- Measure P1 wave latency and amplitude as indicators of visual function [130]
Protein Aggregate Clearance Assessment:
- Filter Trap Assay for Insoluble Aggregates:
- Prepare cell lysates in SDS-containing buffer
- Filter through cellulose acetate membrane under vacuum
- Wash with 0.1% SDS to remove soluble proteins
- Immunodetection of retained aggregates with disease-specific antibodies
- Sarkosyl Insolubility Assay:
- Extract proteins with sarkosyl-containing buffer
- Separate soluble and insoluble fractions by ultracentrifugation
- Analyze fractions by immunoblotting for aggregated proteins
Integrated Signaling Pathways in Proteostasis Regulation
The UPS and autophagy do not operate in isolation but engage in extensive crosstalk and reciprocal regulation. Understanding these interconnected networks is essential for developing effective therapeutic strategies.
Diagram 1: UPS and Autophagy Signaling Pathways. This integrated pathway illustrates the molecular components and regulatory interactions between the ubiquitin-proteasome system (blue) and autophagy pathway (green). The BAG-instructed proteasomal to autophagosomal switch (BIPASS) mechanism (red) enables transition between degradation systems during proteotoxic stress. Chronic failure of both systems leads to apoptotic signaling and neuronal death (gray).
The coordination between UPS and autophagy is mediated by several key mechanisms:
Ubiquitin Signaling Crosstalk: Both systems utilize ubiquitin as a recognition signal, albeit with different chain topologies. K48-linked chains typically target substrates to the proteasome, while K63-linked chains often direct cargo to autophagic degradation [127] [131]. Ubiquitin-binding adaptors such as p62/SQSTM1 recognize polyubiquitinated proteins and deliver them to autophagosomes, providing a direct molecular link between the two systems [127].
Transcription Factor Coordination: Transcription factors such as TFEB and NRF2 regulate both UPS and autophagy components. TFEB is a master regulator of lysosomal biogenesis that also modulates proteasome subunit expression, while NRF2 activation enhances proteasome activity and may influence autophagic processes [129].
BIPASS Mechanism: The "BAG-instructed proteasomal to autophagosomal switch and sorting" represents a critical regulatory node. Under basal conditions, the co-chaperone BAG1 directs ubiquitinated substrates to the proteasome. During cellular stress, BAG3 expression increases and redirects misfolded proteins to autophagy via interactions with HDAC6 and p62, providing a dynamic switch between degradation pathways [130].
UPR-Mediated Regulation: The unfolded protein response (UPR), activated by endoplasmic reticulum stress, coordinates both UPS and autophagy through its three branches (PERK, IRE1α, ATF6). The IRE1α-XBP1 axis upregulates ER-associated degradation (ERAD) components, while the PERK-ATF4 pathway induces autophagy-related genes [129] [132].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for UPS and Autophagy Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| UPS Inhibitors | MG132, Bortezomib, Ixazomib, Epoxomicin | Inhibit proteasome activity; experimental models of UPS impairment | Varying specificity for β5, β1, β2 subunits; different blood-brain barrier penetration |
| Autophagy Inducers | Rapamycin (mTOR-dependent), Trehalose, Carbamazepine (mTOR-independent) | Activate autophagy initiation or flux | Mechanism-dependent effects; mTOR-independent inducers avoid immunosuppression |
| Autophagy Inhibitors | Bafilomycin A1, Chloroquine (late stage), 3-Methyladenine (early stage) | Block autophagosome-lysosome fusion or autophagosome formation | Stage-specific inhibition enables mechanistic dissection |
| UPS Activity Reporters | GFPu, RFPu, Proteasome activity probes | Monitor UPS function in live cells or lysates | Fluorogenic substrates available for different proteasome active sites |
| Autophagy Flux Reporters | GFP-LC3, mRFP-GFP-LC3, LC3B antibodies | Visualize and quantify autophagosomes and autolysosomes | Tandem fluorescent constructs differentiate autophagic compartments |
| E3 Ligase Modulators | Compound 056 (HECT-E3 inhibitor), MLN4924 (NEDD8-activating enzyme inhibitor) | Target specific ubiquitination pathways | Compound 056 shows neuroprotection in optic neuropathy models [130] |
| Ubiquitin Binding Reagents | Ubiquitin antibodies, TUBE (Tandem Ubiquitin Binding Entity) reagents | Detect and purify ubiquitinated proteins | K48- and K63-linkage specific antibodies available |
| LysoTrackers | LysoTracker Red, LysoSensor dyes | Label and monitor lysosomal number, volume, and pH | Useful for assessing lysosomal function in autophagy |
| UPR Modulators | STF-083010 (IRE1α inhibitor), GSK2606414 (PERK inhibitor) | Target specific unfolded protein response pathways | IRE1α inhibition shows efficacy in multiple myeloma models [132] |
| Apoptosis Assays | Annexin V/PI staining, Caspase-3 cleavage antibodies | Assess cell death endpoints | Critical for evaluating therapeutic windows |
The comparative analysis of UPS enhancement versus autophagy activation reveals a complex therapeutic landscape with neither approach demonstrating universal superiority. Instead, efficacy is highly context-dependent, influenced by disease type, stage of pathology, and the specific protein aggregates involved.
Autophagy activation generally shows advantages for clearing large, insoluble aggregates that exceed the physical capacity of the proteasome. This approach has demonstrated significant promise in models of AD, PD, and HD, where large protein aggregates are hallmark pathologies [125] [126]. However, chronic or excessive autophagy induction may lead to autophagic cell death or deplete essential cellular components.
UPS enhancement offers more precise degradation of soluble misfolded proteins and critical regulatory factors. The specificity afforded by the extensive E3 ligase family provides opportunities for targeted interventions, as demonstrated by HECT-E3 inhibition in traumatic optic neuropathy [130]. However, UPS enhancement faces challenges from the physical limitation of the proteasome chamber and the vulnerability of UPS components to inhibition by protein aggregates.
Future therapeutic development should prioritize several key areas:
- Disease-Stratified Approaches: Matching degradation strategies to specific proteinopathies based on aggregate size, solubility, and cellular localization
- Sequential and Combinatorial Strategies: Implementing UPS enhancement followed by autophagy activation or developing compounds that simultaneously modulate both pathways
- Temporal Considerations: Aligning interventions with disease stages, as proteostasis network efficacy declines with aging and progression
- Precision Targeting: Developing E3 ligase-specific modulators to enhance degradation of pathological proteins while sparing normal cellular functions
- Delivery Challenges: Addressing the significant hurdle of blood-brain barrier penetration for central nervous system applications
The most promising emerging paradigm recognizes the interconnected nature of cellular proteostasis. Rather than viewing UPS enhancement and autophagy activation as competing strategies, future interventions should harness their complementary functions, potentially through the BIPASS mechanism or coordinated transcription factor activation. Such integrated approaches offer the greatest potential for addressing the complex challenge of protein misfolding and aggregation in neurodegenerative diseases.
Biomarker Development for Monitoring Therapeutic Response and Disease Progression
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD), represent a growing global health crisis characterized by progressive neuronal loss and cognitive or motor function decline. These disorders share a common pathological hallmark: the misfolding, aggregation, and accumulation of specific proteins that disrupt cellular proteostasis [9]. Pathogenic proteins such as amyloid-β (Aβ), tau, α-synuclein, and TDP-43 adopt β-sheet-rich conformations and form cytotoxic oligomers, protofibrils, and ultimately insoluble fibrils that contribute to the formation of characteristic inclusions like amyloid plaques, neurofibrillary tangles, and Lewy bodies [9]. The development of reliable biomarkers to monitor both disease progression and therapeutic response has therefore become a critical priority in neuroscience research and clinical practice, particularly as targeted therapies against misfolded proteins advance through clinical development.
The global biomarker market reflects this imperative, with the blood-based biomarkers segment projected to grow from USD 8.17 billion in 2025 to USD 15.3 billion by 2035, demonstrating a compound annual growth rate (CAGR) of 6.5% [133]. Neurological disease applications constitute approximately 20% of this market, supported by escalating demands for Alzheimer's disease detection, neurodegenerative disorder monitoring, and brain injury assessment [133]. This growth is fueled by the critical need for non-invasive diagnostic solutions that can detect pathological changes before clinical symptoms emerge, track disease progression with high sensitivity, and quantify response to disease-modifying therapies with precision and reliability.
Biomarker Classes and Their Pathophysiological Basis in Protein Misfolding Disorders
Core Biomarker Categories in Neurodegeneration
Biomarkers for neurodegenerative diseases can be categorized based on their biological basis and clinical application. Understanding these categories is essential for developing a comprehensive monitoring strategy.
Table 1: Core Biomarker Classes in Neurodegenerative Diseases
| Biomarker Class | Key Analytes | Biological Significance | Therapeutic Monitoring Utility |
|---|---|---|---|
| Protein Misfolding/Aggregation | Aβ42/40 ratio, p-tau (181, 217, 231), α-synuclein | Directly measures core pathology of protein misfolding and aggregation [9] | Direct target engagement; reduction indicates pathogen clearance |
| Neuronal Injury/Degeneration | Neurofilament Light (NfL), SNAP-25 | Reflects axonal damage and general neurodegeneration [134] | Monitors downstream protective effects of therapeutics |
| Neuroinflammation | GFAP, YKL-40, various cytokines | Indicates glial activation and inflammatory responses to protein aggregates [9] | Assesses modulation of neuroinflammatory pathways |
| Synaptic Dysfunction | Synaptotagmin, SNAP-25, neurogranin | Measures synaptic integrity and function [134] | Evaluates functional recovery and connectivity preservation |
Blood-Based Biomarkers: Performance Characteristics and Clinical Validation
Blood-based biomarkers (BBMs) represent a transformative advancement in neurodegenerative disease monitoring, offering less invasive, more accessible, and cost-effective alternatives to cerebrospinal fluid (CSF) analysis or PET imaging. The Alzheimer's Association has established evidence-based clinical practice guidelines specifying that BBM tests with ≥90% sensitivity and ≥75% specificity can be used as triaging tests, while those with ≥90% sensitivity and specificity can serve as substitutes for amyloid PET imaging or CSF AD biomarker testing in specialized care settings [135].
Key plasma biomarkers demonstrating clinical utility include:
- Plasma phosphorylated-tau (p-tau) variants: p-tau217, p-tau181, and p-tau231 show strong correlation with amyloid and tau pathology, with p-tau217 exhibiting particularly high diagnostic accuracy for Alzheimer's disease [135] [134].
- Amyloid-β42/40 ratio: This ratio demonstrates high sensitivity and specificity for detecting cerebral amyloidosis, with decreasing ratios reflecting increasing amyloid burden [135].
- Neurofilament Light (NfL): As a marker of neuroaxonal injury, NfL elevations occur across multiple neurodegenerative conditions and effectively track disease progression [134].
- Glial Fibrillary Acidic Protein (GFAP): This marker of astrocytic activation shows particular utility in detecting early Alzheimer's pathology and inflammatory responses [134].
Technological advancements in analytical sensitivity have been crucial for realizing the potential of BBMs. Platforms such as Simoa (Single Molecule Array) technology provide up to 1000-fold greater sensitivity than conventional immunoassays, enabling detection of central nervous system biomarkers in blood that were previously measurable only in CSF [134]. Emerging approaches include dried blood spot kits that further enhance accessibility and facilitate large-scale population screening [134].
Methodologies and Experimental Protocols for Biomarker Analysis
Analytical Platforms for Biomarker Detection
Ultra-sensitive immunoassay platforms form the cornerstone of modern biomarker analysis for neurodegenerative diseases. The following protocol outlines a standardized approach for quantifying key biomarkers in plasma samples:
Protocol 1: Multiplex Biomarker Analysis in Plasma Using Simoa Technology
Sample Collection and Preparation: Collect blood into EDTA tubes and centrifuge at 2,000×g for 10 minutes at 4°C. Aliquot plasma and store at -80°C until analysis. Avoid freeze-thaw cycles.
Reagent Preparation: Reconstitute lyophilized calibrators and quality control materials according to manufacturer specifications. Prepare detector antibodies, streptavidin-β-galactosidase (SBG), and resorufin-β-D-galactopyranoside (RGP) substrate according to kit protocols.
Assay Procedure:
- Pipette 100µL of plasma (diluted 1:4) into sample plates.
- Add biotinylated capture antibodies and paramagnetic beads coated with secondary antibodies.
- Incubate with shaking (500rpm) for 30 minutes at room temperature.
- Transfer beads to wash plates and perform three washes with manufacturer's wash buffer.
- Incubate with SBG conjugate for 5 minutes, followed by three additional washes.
- Resuspend beads in RGP substrate solution and load into the Simoa HD-X Analyzer.
Data Analysis: Generate standard curves using a four-parameter logistic (4-PL) fit model. Calculate analyte concentrations from the average enzyme per bead (AEB) values. Apply dilution factors for final concentration reporting.
This methodology enables simultaneous quantification of multiple biomarkers, including p-tau species, NfL, GFAP, and SNAP-25, with femtomolar sensitivity [134]. The exceptional sensitivity of this approach allows detection of central nervous system-derived proteins in blood despite their low abundance and the interference from high-abundance plasma proteins.
Biomarker Validation in Clinical Trials
Robust biomarker validation is essential for establishing clinical utility in therapeutic monitoring. The PRECISE-AD Phase 1b trial of PMN310 (an anti-AβO antibody) provides an exemplary model for comprehensive biomarker integration:
Protocol 2: Biomarker Assessment in Clinical Trials of Disease-Modifying Therapies
Primary Safety Endpoints:
- Regular MRI monitoring for amyloid-related imaging abnormalities (ARIA) [104]
- Standard safety laboratories, vital signs, and adverse event recording
Pharmacodynamic Biomarkers:
- Plasma Aβ oligomer levels to demonstrate target engagement
- Aβ42/40 ratio to assess effects on amyloid processing
- p-tau species (p-tau181, p-tau217) as indicators of downstream tau pathology
Disease Progression Markers:
- Neurofilament Light (NfL) to monitor neuroaxonal injury
- GFAP for tracking glial activation and inflammatory responses
- Cognitive assessments (e.g., CDR-SB, ADAS-Cog) for clinical correlation
Sampling Schedule:
- Baseline, 3-month, 6-month, and 12-month intervals for comprehensive trajectory analysis
- Blinded interim analyses at 6 months to assess biomarker trends while maintaining trial integrity [104]
This multidimensional approach enables comprehensive assessment of both biological target engagement and clinical efficacy, providing critical insights into therapeutic mechanisms and treatment effects.
Quantitative Biomarker Data: Market Landscape and Performance Metrics
The accelerating development of biomarkers for neurodegenerative diseases is reflected in both market growth and analytical performance improvements. Quantitative data provides critical context for understanding the landscape and potential of these tools.
Table 2: Blood-Based Biomarkers Market Forecast and Application (2025-2035)
| Segment | 2025 Value (USD Billion) | 2035 Projected Value (USD Billion) | CAGR | Market Share (2025) |
|---|---|---|---|---|
| Genetic Biomarkers | 2.77 | 5.19 | 6.5% | 33.9% |
| Protein Biomarkers | 1.67 | 3.14 | 6.5% | 20.5% |
| Cell-based Biomarkers | 1.47 | 2.75 | 6.5% | 18.0% |
| Cancer Applications | 3.17 | 5.93 | 6.5% | 38.8% |
| Neurological Applications | 1.63 | 3.06 | 6.5% | 20.0% |
| Cardiovascular Applications | 2.04 | 3.83 | 6.5% | 25.0% |
Data sourced from FactMR Blood Based Biomarkers Market report [133]
The market data reveals several key insights: genetic biomarkers command the largest market share (33.9%) due to their essential role in precision medicine and hereditary risk assessment, while neurological applications constitute a substantial and growing segment (20.0%) [133]. The projection of consistent growth across all segments underscores the expanding role of biomarkers in healthcare and clinical research.
Table 3: Diagnostic Performance of Key Alzheimer's Blood-Based Biomarkers
| Biomarker | Sensitivity | Specificity | Primary Utility | Technology Platform |
|---|---|---|---|---|
| p-tau217 | 92-97% | 93-98% | Alzheimer's diagnosis and differential diagnosis [135] | Mass spectrometry, immunoassays |
| p-tau181 | 90-94% | 88-93% | Alzheimer's diagnosis and progression [135] | Immunoassays |
| Aβ42/40 ratio | 89-95% | 85-92% | Detection of amyloid pathology [135] | Immunoassays |
| GFAP | 85-90% | 82-88% | Astrocyte activation, early pathology [134] | Immunoassays |
| NfL | 80-87% | 78-85% | Neuroaxonal injury, disease progression [134] | Immunoassays |
Performance characteristics based on Alzheimer's Association Clinical Practice Guidelines and recent validation studies [135] [134]
The performance metrics in Table 3 demonstrate that several blood-based biomarkers now meet or exceed the Alzheimer's Association recommended thresholds for clinical use (≥90% sensitivity, ≥75% specificity for triaging; ≥90% sensitivity and specificity for substitution of PET/CSF) [135]. This represents a significant advancement from just a few years ago when BBMs were considered primarily research tools.
Signaling Pathways and Experimental Workflows
The molecular pathways connecting protein misfolding to neurodegeneration provide critical context for biomarker interpretation and therapeutic targeting. The following diagram illustrates key pathological mechanisms and corresponding biomarker responses.
Diagram 1: Protein Misfolding Pathways and Corresponding Biomarkers (Width: 760px)
This pathway illustration demonstrates the cascade from initial protein misfolding to clinical symptoms, highlighting points where specific biomarkers provide windows into pathological processes. The dashed lines indicate biomarker-pathology relationships essential for monitoring disease progression and therapeutic response.
The implementation of biomarker development follows a systematic workflow from discovery to clinical application, as detailed in the following diagram:
Diagram 2: Biomarker Development and Validation Workflow (Width: 760px)
This workflow illustrates the structured process required to translate potential biomarkers from discovery to clinical application, highlighting essential technologies and methodologies at each stage. The progression from basic research to clinical practice guidelines ensures robust validation and appropriate implementation.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful biomarker development and implementation requires specialized reagents, platforms, and analytical tools. The following table details essential components of the modern biomarker researcher's toolkit.
Table 4: Essential Research Reagents and Platforms for Biomarker Development
| Tool Category | Specific Products/Platforms | Primary Function | Key Applications |
|---|---|---|---|
| Ultra-Sensitive Detection Platforms | Simoa HD-X Analyzer, MSD S-PLEX, Ella | Femtogram-level detection of low-abundance biomarkers in blood [134] | Quantifying CNS-derived proteins in blood; therapeutic monitoring |
| Protein Aggregation Assays | PET tracers (florbetapir, flortaucipir), conformation-specific antibodies | Detection and quantification of misfolded protein aggregates [136] | Target engagement assessment for anti-aggregation therapies |
| Reference Materials | Certified reference standards for Aβ42, Aβ40, p-tau variants | Assay calibration and standardization across laboratories [135] | Ensuring reproducibility and comparability across sites |
| Sample Collection Systems | Dried blood spot kits, specialized blood collection tubes | Standardized sample acquisition and stabilization [134] | Multi-site trials; remote sampling; biobanking |
| Data Analysis Software | R/Bioconductor packages, Simoa Data Analysis Software | Statistical analysis and interpretation of biomarker data [133] | Modeling biomarker trajectories; clinical endpoint correlation |
This toolkit enables researchers to address the significant technical challenges in biomarker development, particularly the need to detect extremely low concentrations of CNS-derived proteins in peripheral blood and distinguish pathological protein conformations from normal variants.
Biomarker development for monitoring therapeutic response and disease progression in neurodegenerative diseases represents a rapidly advancing field that is transforming both clinical trial design and patient care. The integration of ultra-sensitive detection platforms with validated biomarker panels creates unprecedented opportunities to track the underlying pathology of protein misfolding disorders and quantify response to disease-modifying therapies. Blood-based biomarkers in particular have reached a stage of maturity where they can now be deployed in clinical trials and specialized care settings with confidence in their analytical and clinical performance [135].
Future directions in the field include the development of multi-analyte panels that combine markers of different pathological processes (protein aggregation, synaptic dysfunction, neuroinflammation) to provide comprehensive profiles of disease state and progression [9]. The integration of artificial intelligence and machine learning approaches will enhance the predictive value of biomarker data, enabling more precise prognosis and personalized treatment selection [133] [137]. Additionally, the validation of biomarkers in diverse populations will be essential to ensure equitable application of emerging therapies across different demographic and genetic backgrounds [134].
As targeted therapies against protein misfolding and aggregation advance through clinical development, including antibodies such as PMN310 that selectively target toxic oligomers [104], the role of biomarkers will continue to expand. The systematic implementation of validated biomarkers in clinical trials and practice will accelerate the development of effective treatments and ultimately enable a precision medicine approach to neurodegenerative diseases that has remained elusive until now. Through continued refinement of analytical methods and rigorous clinical validation, biomarkers will play an indispensable role in defeating the growing challenge of neurodegenerative disorders.
Conclusion
The field of protein misfolding in neurodegenerative diseases has evolved from recognizing pathological aggregates to understanding dynamic proteostasis networks and developing sophisticated intervention strategies. The integration of AI-driven prediction platforms with traditional experimental methods represents a paradigm shift in early-stage therapeutic development, enabling proactive rather than reactive approaches. Future directions must focus on translating mechanistic insights into clinically effective therapies, particularly through personalized medicine approaches that account for individual proteostatic capacities. The development of sensitive biomarkers for early detection and therapeutic monitoring, combined with multi-target strategies addressing different stages of the aggregation cascade, will be crucial for meaningful clinical advancements. Collaborative ecosystems engaging patients, clinicians, researchers, and regulatory agencies will accelerate the progression from fundamental discovery to transformative patient treatments.
References
- 1. Navigating the landscape of protein folding and proteostasis [https://www.nature.com/articles/s41392-025-02439-w]
- 2. Targeting proteostasis for cancer therapy: current advances ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02472-x]
- 3. The Proteostasis Network in Proteinopathies - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12597693/]
- 4. Quantitative analysis of proteostasis networks [https://pmc.ncbi.nlm.nih.gov/articles/PMC10928379/]
- 5. The endoplasmic reticulum proteostasis network profoundly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8906867/]
- 6. Proteostasis signatures in human diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12173376/]
- 7. The Global Neurodegeneration Proteomics Consortium [https://www.nature.com/articles/s41591-025-03834-0]
- 8. Current Understanding of Protein Aggregation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12610759/]
- 9. Exploring Protein Misfolding and Aggregate Pathology in ... [https://www.mdpi.com/2076-3417/15/18/10285]
- 10. Protein misfolding in neurodegenerative diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC5348787/]
- 11. Misfolding and aggregation in neurodegenerative diseases [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-024-01791-8]
- 12. Misfolded proteins and neurodegenerative diseases [https://www.bmglabtech.com/en/blog/misfolded-proteins-and-neurodegenerative-diseases/]
- 13. 综述:阿尔茨海默病中的TDP-43:病理生理学与治疗策略 [https://www.ebiotrade.com/newsf/2025-10/20251003031817157.htm]
- 14. Blood Biomarkers in Neurodegenerative Diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC10435056/]
- 15. Biomarkers and therapeutic strategies targeting microglia in ... [https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-025-00867-4]
- 16. Current Understanding of Protein Aggregation in ... [https://pubmed.ncbi.nlm.nih.gov/41226604/]
- 17. Current Understanding of Protein Aggregation in ... [https://www.mdpi.com/1422-0067/26/21/10568]
- 18. “Prion-like” seeding and propagation of oligomeric protein ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2024.1436262/full]
- 19. Key Steps for α-Synuclein Prion-Like Propagation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4276803/]
- 20. Label-free identification of protein aggregates using deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10684545/]
- 21. Self-driving microscopy detects the onset of protein ... [https://www.nature.com/articles/s41467-025-60912-0]
- 22. The proteasome as a druggable target with multiple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7236745/]
- 23. The Role of Ubiquitin-Proteasome Pathway and Autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7016479/]
- 24. Mechanisms of Protein Degradation [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/protein-degradation-resource-center/mechanisms-protein-degradation.html]
- 25. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 26. Review Autophagy and the ubiquitin-proteasome system [https://www.sciencedirect.com/science/article/pii/S0925443908001944]
- 27. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 28. Advances in the structures, mechanisms and targeting of ... [https://www.nature.com/articles/s41392-025-02166-2]
- 29. Protein Quality Control by Molecular Chaperones in ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2017.00185/full]
- 30. Proteotoxic stress and inducible chaperone networks in ... [https://genesdev.cshlp.org/content/22/11/1427]
- 31. Molecular Chaperone Dysfunction in Neurodegenerative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4217372/]
- 32. Mechanistic insights of molecular chaperone Hsp70 [https://www.sciencedirect.com/science/article/pii/S3050623925000513]
- 33. Emerging opportunities and challenges in small molecule ... [https://pubmed.ncbi.nlm.nih.gov/40907379/]
- 34. Prion-like Mechanisms in Neurodegenerative Diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3648341/]
- 35. From the prion-like propagation hypothesis to therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6942016/]
- 36. Protein misfolding in neurodegenerative diseases [https://translationalneurodegeneration.biomedcentral.com/articles/10.1186/s40035-017-0077-5]
- 37. Prion-like propagation of human brain-derived alpha ... [https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-015-0254-7]
- 38. What is the evidence that tau pathology spreads through ... [https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-017-0488-7]
- 39. Excitatory neuron-prone prion propagation ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11669680/]
- 40. Protein aggregation in neurodegenerative diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574514/]
- 41. AggNet: Advancing protein aggregation analysis through ... [https://pubmed.ncbi.nlm.nih.gov/39840791/]
- 42. Computational methods to predict protein aggregation [https://www.sciencedirect.com/science/article/pii/S0959440X22000161]
- 43. Enhancing protein aggregation prediction: a unified ... [https://pubs.rsc.org/en/content/articlelanding/2024/ra/d4ra06285j]
- 44. AI tool unlocks long-standing biomedical mystery behind ... [https://news.rice.edu/news/2025/ai-tool-unlocks-long-standing-biomedical-mystery-behind-alzheimers-parkinsons]
- 45. AlphaFold Protein Structure Database [https://alphafold.ebi.ac.uk/]
- 46. Study Reveals an Unexpected Role for Protein Aggregates ... [https://www.massgeneralbrigham.org/en/about/newsroom/articles/new-study-links-protein-aggregation-to-brain-disease]
- 47. Misfolded protein oligomers: mechanisms of formation ... [https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-023-00651-2]
- 48. A rapid solvent estimator for accessible ... surface area coarse grained [https://pubmed.ncbi.nlm.nih.gov/28419507/]
- 49. sciencedirect.com/topics/engineering/ coarse - grained - molecular ... [https://www.sciencedirect.com/topics/engineering/coarse-grained-molecular-dynamic]
- 50. A Standardized Benchmark for Machine-Learned ... [https://arxiv.org/html/2510.17187v1]
- 51. Force field - GROMACS 2025.3 documentation [https://manual.gromacs.org/documentation/2025.3/reference-manual/functions/force-field.html]
- 52. Combining molecular dynamics simulations and ... [https://pubmed.ncbi.nlm.nih.gov/31928730/]
- 53. β-Amyloid and the Pathomechanisms of Alzheimer's Disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC6151811/]
- 54. Integrative structural modeling with small angle X-ray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3427135/]
- 55. Artificial intelligence-driven approaches for the rational ... [https://www.nature.com/articles/s44431-025-00005-6]
- 56. Reinforcement Learning-Based Target-Specific De Novo ... [https://pubmed.ncbi.nlm.nih.gov/40824037/]
- 57. Combining genetic algorithm with machine learning strategies ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04156-x]
- 58. De Novo Design of Specific Heterotrimeric Collagen‐Like ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12533406/]
- 59. Populating Chemical Space with Peptides Using a Genetic ... [https://pubmed.ncbi.nlm.nih.gov/31868369/]
- 60. Design of target specific peptide inhibitors using generative ... [https://www.nature.com/articles/s41467-024-45766-2]
- 61. Mathematical Modeling of Protein Misfolding Mechanisms ... [https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2018.00037/full]
- 62. Classic and contemporary approaches to modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2932968/]
- 63. Development of a Simple Kinetic Mathematical Model of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7345685/]
- 64. How and why to build a mathematical model: A case study ... [https://www.sciencedirect.com/science/article/pii/S0021925817485966]
- 65. How One Scientist's Misfolded Protein Breakthrough is Saving ... [https://wewillcure.com/insights/neurology/therapeutics/how-one-scientists-misfolded-protein-breakthrough-is-saving-lives]
- 66. Targeting protein misfolding in Alzheimer's disease [https://www.sciencedirect.com/science/article/pii/S0753332225007255]
- 67. Challenges and limitations in computational prediction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10796696/]
- 68. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 69. Integrating AlphaFold2 models and clinical data to improve ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12338786/]
- 70. Integrating AlphaFold2 models and clinical data to improve ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012829]
- 71. The Role of AlphaFold in Solving Protein Misfolding ... [https://gaintherapeutics.com/perspectives/why-has-alphafold-protein-structure-database-become-so-important-for-protein-misfolding/]
- 72. Protein misfolding: understanding biology to classify and ... [https://pubmed.ncbi.nlm.nih.gov/39932548/]
- 73. Protein misfolding that propagates and the mechanisms of ... [https://neuroscience.uchicago.edu/protein-misfolding-propagates-and-mechanisms-neurodegenerative-disease]
- 74. The Future Of Biologics Delivery High-Concentration ... [https://www.drugdeliveryleader.com/doc/the-future-of-biologics-delivery-high-concentration-subcutaneous-injectables-0001]
- 75. Development roadmap for subcutaneous delivery of high ... [https://pubmed.ncbi.nlm.nih.gov/40706801/]
- 76. Advanced Strategies for High-Concentration Protein ... [https://www.coriolis-pharma.com/advanced-strategies-for-high-concentration-protein-formulations/]
- 77. Current and emerging strategies for subcutaneous delivery ... [https://pubmed.ncbi.nlm.nih.gov/40516697/]
- 78. Advanced Formulations/Drug Delivery Systems for ... [https://pubmed.ncbi.nlm.nih.gov/36058255/]
- 79. The Role of Functional Excipients in Solid Oral Dosage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7284856/]
- 80. Protein Aggregation - Neurodegeneration - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK208522/]
- 81. A Review on the Stability Challenges of Advanced Biologic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114724/]
- 82. Factors Affecting the Pharmacology of Antibody–Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6698819/]
- 83. Optimizing and Intensifying ADC Aggregate Removal [https://www.bioprocessintl.com/process-development/optimizing-and-intensifying-adc-aggregate-removal-a-doe-approach-to-membrane-chromatography-and-rapid-cycling]
- 84. A comprehensive review of key factors affecting the efficacy ... [https://www.sciencedirect.com/science/article/pii/S0753332223001968]
- 85. Progress and challenges in viral vector manufacturing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4802372/]
- 86. Proteostasis and Its Role in Disease Development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123047/]
- 87. Proteostasis and Unfolded Protein Response Dynamics in ... [https://pubmed.ncbi.nlm.nih.gov/40832271/]
- 88. Chaperone‐mediated autophagy: Molecular mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10466100/]
- 89. Chaperone Mediated Autophagy in the Crosstalk of ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2018.00778/full]
- 90. Proteostasis signatures in human diseases - Research journals [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013155]
- 91. Regulated degradation of Chk1 by chaperone-mediated ... [https://www.nature.com/articles/ncomms7823]
- 92. Cross-seeding of alpha-synuclein aggregation by amyloid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7949133/]
- 93. α-synuclein and tau: interactions, cross-seeding, and the ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2025.1570553/full]
- 94. Immunization targeting diseased proteins in synucleinopathy ... [https://translationalneurodegeneration.biomedcentral.com/articles/10.1186/s40035-025-00490-9]
- 95. Therapeutic Protein Aggregation: Mechanisms, Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4146573/]
- 96. Therapeutic Approaches Targeting Protein Aggregation in ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2020.00098/full]
- 97. Mechanisms of Aβ Clearance and Degradation by Glial Cells [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2016.00160/full]
- 98. Amyloid-Beta Protein Clearance and Degradation (ABCD ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4820400/]
- 99. 'Death fold' proteins can make cells self-destruct. Scientists ... [https://www.npr.org/sections/shots-health-news/2025/10/16/nx-s1-5575582/death-fold-proteins-apoptosis-alzheimers-cancer]
- 100. Computational Mechanisms of Protein Misfolding [https://www.mdpi.com/1422-0067/26/22/11021]
- 101. Protein aggregation and neurodegenerative disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC10834863/]
- 102. Prediction and evaluation of protein aggregation with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12126135/]
- 103. A multi-dimensional Structure-Activity Relationship ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3004770/]
- 104. ProMIS Neurosciences Announces Second Quarter 2025 ... [https://www.promisneurosciences.com/news-media/press-releases/detail/251/promis-neurosciences-announces-second-quarter-2025]
- 105. Nano-conjugation of small molecule modulators of protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025088506]
- 106. Conformational inhibitors of protein aggregation [https://www.sciencedirect.com/science/article/abs/pii/S0959440X23001744]
- 107. Emerging Alzheimer's Therapies: AD/PD 2025 Conference [https://www.delveinsight.com/blog/ad-pd-2025-conference-highlights]
- 108. Plant-Based Inhibitors of Protein Aggregation [https://pubmed.ncbi.nlm.nih.gov/40305223/]
- 109. Protein misfolding: understanding biology to classify and treat ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12630200/]
- 110. Antibodies and protein misfolding: From structural research ... [https://www.sciencedirect.com/science/article/pii/S1570963914002222]
- 111. Targeting protein misfolding in Alzheimer's disease [https://pubmed.ncbi.nlm.nih.gov/40925230/]
- 112. Production of Monoclonal Antibodies to Pathologic β-sheet ... [https://www.nature.com/articles/s41598-017-10393-z]
- 113. Antibody Aggregation: Insights from Sequence and Structure [https://pmc.ncbi.nlm.nih.gov/articles/PMC6698864/]
- 114. Anti-Amyloid Monoclonal Antibodies for Alzheimer's Disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC12470750/]
- 115. Akeso's Bispecific Antibody Targeting Aβ and BBB- ... [https://www.akesobio.com/en/media/akeso-news/251117/]
- 116. Data-driven modeling of amyloid-β targeted antibodies for ... [https://www.nature.com/articles/s41540-025-00610-1]
- 117. Anti-Drug Antibody Response to Therapeutic ... [https://www.mdpi.com/2227-9059/13/2/299]
- 118. Review Gene therapy strategies for aging intervention [https://www.sciencedirect.com/science/article/pii/S2772892725000288]
- 119. First Synthetic 'Mini Prion' Shows How Protein Misfolding ... [https://news.weinberg.northwestern.edu/2025/04/28/first-synthetic-mini-prion-shows-how-protein-misfolding-multiplies/]
- 120. Protein aggregation and its affecting mechanisms in ... [https://pubmed.ncbi.nlm.nih.gov/39396709/]
- 121. Editorial: Molecular Chaperones and Neurodegeneration [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2017.00565/full]
- 122. Modulation of Heat Shock Proteins Levels in Health and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248682/]
- 123. Quantitative Comparison of HSF1 Activators - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9259536/]
- 124. First complete structures of heat shock chaperone protein ... [https://www.eurekalert.org/news-releases/1101842]
- 125. Autophagy in neurodegenerative diseases: pathogenesis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5739982/]
- 126. Degradation of misfolded proteins in neurodegenerative ... [https://www.nature.com/articles/emm2014117]
- 127. Crosstalk Between Mammalian Autophagy and the Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6175981/]
- 128. Ubiquitin-Proteasome System in Neurodegenerative Disorders [https://pmc.ncbi.nlm.nih.gov/articles/PMC6370320/]
- 129. Ubiquitination-Proteasome System (UPS) and Autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8909305/]
- 130. Tailoring UPS-to-autophagy transition to restore RGC ... [https://www.sciencedirect.com/science/article/abs/pii/S001448352500260X]
- 131. The ubiquitin-proteasome system in neurodegenerative ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2014.00070/full]
- 132. Targeting autophagy increases the efficacy of proteasome ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-022-09775-y]
- 133. Blood Based Biomarkers Market [https://www.factmr.com/report/blood-based-biomarkers-market]
- 134. Key Takeaways from AD/PD 2025 [https://www.quanterix.com/blog-driving-precision-in-neurology-key-takeaways-from-ad-pd-2025/]
- 135. Alzheimer's Association Clinical Practice Guideline on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306682/]
- 136. 2025 NIH Alzheimer's Disease and Related Dementias ... [https://www.nia.nih.gov/about/2025-nih-dementia-research-progress-report]
- 137. Analysis and Forecasts 2025-2033 [https://www.datainsightsmarket.com/reports/biomarker-1471572]