Hsp70 and Hsp60 Chaperone Mechanisms: Orchestrating Proteostasis in Health and Disease
This article provides a comprehensive analysis of the structural and functional mechanisms of Hsp70 and Hsp60 molecular chaperones in maintaining cellular proteostasis.
Hsp70 and Hsp60 Chaperone Mechanisms: Orchestrating Proteostasis in Health and Disease
Abstract
This article provides a comprehensive analysis of the structural and functional mechanisms of Hsp70 and Hsp60 molecular chaperones in maintaining cellular proteostasis. We explore the foundational biology of these chaperone systems, including their ATP-dependent allosteric regulation and complex cooperation with co-chaperones. The review examines cutting-edge methodological approaches for investigating chaperone functions and discusses the direct implications of chaperone dysregulation in human pathologies, including cancer and neurodegenerative diseases. A significant focus is placed on emerging therapeutic strategies that target these chaperone networks, highlighting small-molecule inhibitors and innovative drug design approaches currently in development. This synthesis of mechanistic insights and translational applications offers researchers and drug development professionals a strategic framework for advancing chaperone-targeted therapeutics.
Structural Biology and Core Mechanisms of Hsp70 and Hsp60 Chaperone Systems
Evolutionary Conservation and Genomic Organization of Chaperone Families
Molecular chaperones, including the highly conserved Hsp70 and Hsp60 families, constitute fundamental components of the cellular protein homeostasis (proteostasis) network. These proteins facilitate nascent polypeptide folding, prevent protein aggregation, mediate intracellular trafficking, and support stress response pathways. The evolutionary conservation and genomic organization of these chaperone families reflect their critical role in maintaining cellular viability under both normal and stress conditions. Understanding their structural and genomic features provides crucial insights for developing therapeutic interventions for protein misfolding diseases, cancer, and neurodegenerative disorders. This review examines the evolutionary trajectories, structural conservation, and genomic architecture of Hsp70 and Hsp60 chaperone families within the broader context of proteostasis research.
Structural Conservation and Evolutionary Relationships
Hsp60 Chaperonin Family
The Hsp60 family, also known as chaperonin 60 (Cpn60), demonstrates remarkable evolutionary conservation from prokaryotes to eukaryotes. In prokaryotes such as Escherichia coli, Hsp60 functions as the GroEL/GroES complex, forming a double-ring barrel structure that facilitates protein folding within a central cavity [1]. This complex captures non-native substrate proteins through hydrophobic interactions and encloses them in the central ring cavity where folding occurs protected from aggregation [1].
Eukaryotic Hsp60 localizes primarily to mitochondria (80-85%) with the remainder (15-20%) found in the cytoplasm [1]. The protein forms a double-ring assembly with each subunit approximately 60 kDa, functioning as a molecular chaperone for naïve polypeptides and unfolded proteins in both cytoplasmic and mitochondrial compartments [1]. The table below summarizes the key classification and characteristics of chaperonin families:
Table 1: Classification and Characteristics of Chaperonin Families
| Group | Chaperonins | Organisms | Localization | Structural Characteristics |
|---|---|---|---|---|
| Group I | GroEL/GroES, HSP60/HSP10, Cpn60, HSPD1/E | Prokaryotes, Eukaryotes | Cytoplasm, Mitochondria, Nucleus, Chloroplast | Homo-oligomeric, GroES/HSP10-dependent, seven subunits per ring |
| Group II | TRiC/CCT, TCP1 | Archaea, Eukaryotes cytosol | Cytosol | Hetero-oligomeric, GroES/HSP10-independent, eight to nine subunits per ring |
Structural studies reveal that Hsp60 monomers consist of three distinct domains: an apical domain forming the barrel, an intermediate domain connecting the other domains, and an equatorial domain at the base of the ring [1]. The binding and hydrolysis of ATP triggers conformational changes that facilitate substrate protein folding and release. Under pathological conditions, increased Hsp60 can exhibit either pro-survival or lethal functions depending on whether it exists in monomeric or oligomeric forms [1].
Hsp70 Chaperone Family
The Hsp70 family represents one of the most evolutionarily conserved chaperone systems across species. These proteins contain three main domains: an N-terminal nucleotide-binding domain (NBD), a substrate-binding domain (SBD), and a C-terminal substrate-binding region [2]. The NBD exhibits ATPase activity that regulates substrate binding and release cycles, while the SBD interacts with client proteins through hydrophobic interactions.
Hsp70 proteins localize to various cellular compartments including cytoplasm, nucleus, mitochondria, chloroplasts, and endoplasmic reticulum [2]. In plants, the Hsp70 multigene family has expanded significantly, with 113 Hsp70 transcription factors identified in hybrid tea rose (Rosa hybrida), classifying into three phylogenetic subfamilies (I, II, and III) [2]. Most members (51 TFs) fall into subfamily II, exhibiting wide gene structural variations among subfamilies [2].
Genomic Organization and Family Expansion
Evolutionary Expansion of Chaperone Families
Chaperone families have undergone significant expansion throughout evolution, reflecting adaptation to increased proteostatic demands in multicellular organisms. The HSP40 family expanded from three members in E. coli and 22 members in budding yeast to 49 members in humans [3]. Similarly, the evolution of the sHSP family shows complex patterns, with plants and some lower eukaryotes possessing more sHSPs than mammals and higher eukaryotes [3].
This expansion has facilitated functional specialization, with different chaperone isoforms acquiring distinct substrate specificities and regulatory mechanisms. While prokaryotes utilize the same chaperones for both de novo protein folding and stress response, unicellular eukaryotes evolved two separately regulated systems—a basal system and a stress-inducible system—composed of distinct members of the same chaperone families [3]. This two-level organization is conserved in multi-cellular eukaryotes but further complicated by tissue-specific requirements.
Genomic Architecture Across Species
The genomic organization of chaperone genes reveals both conserved and species-specific patterns. In Arabidopsis thaliana, 18 HSP70 genes show varied expression across different tissues, developmental stages, and environmental conditions [2]. Rice (Oryza sativa) possesses 32 HSP70 genes, most exhibiting tissue-specificity and sensitivity to environmental stress [2]. Similarly, soybean (Glycine max) contains 61 HSP70 genes involved in tissue development and response to drought and heat stress [2].
Table 2: HSP70 Gene Family Size Across Species
| Organism | HSP70 Gene Count | Tissue Specificity | Stress Responsiveness |
|---|---|---|---|
| Arabidopsis thaliana | 18 | Varied across tissues | Heat stress responsive |
| Oryza sativa (rice) | 32 | Tissue-specific | Environmental stress sensitive |
| Glycine max (soybean) | 61 | Involved in tissue development | Drought and heat stress responsive |
| Rosa hybrida (tea rose) | 113 | Strong organ specificity | Heat stress responsive |
The hybrid tea rose exemplifies the extensive expansion of HSP70 genes, with 113 identified members showing wide gene structural variations [2]. Group I and II members typically lack introns, while group III members contain 1-4 exons and introns [2]. Promoter analysis of these genes reveals numerous cis-acting elements associated with abiotic stress, hormone response, growth and development, and light response [2].
Tissue-Specific Expression Patterns
Core and Variable Chaperone Networks
Recent transcriptomic analyses across human tissues reveal that the chaperone system is composed of core elements uniformly expressed across tissues and variable elements differentially expressed to meet tissue-specific requirements [3]. Approximately 74% of chaperones are expressed in all human tissues, compared to 47% of other protein-coding genes [3]. Core chaperones are significantly more abundant across tissues and more essential for cell survival than variable chaperones [3].
This layered architecture forms tissue-specific functional networks that are established during development and decline with age [3]. Analysis of human organ development and aging brain transcriptomes reveals that these functional networks are established in development and deteriorate during aging processes [3].
Expression Regulation
Chaperone expression exhibits complex regulation patterns. In hybrid tea rose, gene expression analysis revealed that 57 HSP70 genes displayed strong organ specificity and response to heat stress [2]. Notably, expression levels of RhHSP70-69 and RhHSP70-88 increased significantly after heat stress, suggesting crucial roles in thermotolerance [2]. Subcellular localization confirmed these proteins reside in the nucleus [2].
The quantitative composition of chaperone systems significantly impacts proteostatic capacity. While chaperone overexpression typically enhances folding capacity, specific chaperones when overexpressed can disrupt folding—as demonstrated with FKBP51 overexpression in a tau transgenic mouse model leading to tau accumulation and toxic oligomers [3].
Experimental Approaches for Studying Chaperone Systems
Interaction Mapping Techniques
Advanced proteomic approaches have enabled systematic characterization of chaperone interactions. One comprehensive study combined mass spectrometry and quantitative high-throughput LUMIER assays to map chaperone/co-chaperone/client interaction networks in human cells [4]. This approach identified hundreds of novel chaperone clients and delineated their participation in specific co-chaperone complexes.
The LUMIER method involves stably expressing prey proteins fused to Renilla luciferase in 293T cells. Putative interactors (baits) are tagged with a 3xFLAG epitope and transfected into the reporter cell line. Cell lysates are incubated on anti-FLAG coated plates, and interactions are quantified via luminescence detection [4]. This method enables quantitative assessment of protein-protein interactions with high sensitivity.
Diagram 1: Experimental Workflow for Chaperone Interaction Mapping. This diagram illustrates the integrated approach combining affinity purification mass spectrometry (AP-MS) and LUMIER assays for systematic characterization of chaperone complexes.
Phylogenetic and Genomic Analysis Methods
Phylogenetic analysis of chaperone families typically involves multiple sequence alignment and construction of evolutionary trees using neighbor-joining or maximum likelihood methods. For example, analysis of rose HSP70 genes utilized MEGA7 software with 1000 bootstrap replicates to establish reliable phylogenetic relationships [2].
Genome-wide identification employs tools such as TBtools for preliminary screening, Pfam database searches using hidden Markov models (HMMs), and SMART and NCBI CDD for domain identification [2]. Promoter analysis typically examines 2000 bp upstream of the start codon using platforms like PlantCARE to identify cis-acting elements [2].
Research Reagent Solutions
Table 3: Essential Research Reagents for Chaperone Studies
| Reagent/Tool | Application | Function and Utility |
|---|---|---|
| 3xFLAG-V5 Epitope Tags | Protein interaction studies | Enables affinity purification and identification of chaperone complexes |
| Renilla Luciferase | LUMIER assays | Quantitative measurement of protein-protein interactions |
| SAINT Algorithm | Mass spectrometry data analysis | Statistical framework for identifying high-confidence interactors |
| HMMER/Pfam Databases | Gene family identification | Identification of conserved chaperone domains in genomic sequences |
| TPR Domain Constructs | Co-chaperone studies | Investigation of Hsp90 and Hsp70 interaction mechanisms |
| CDC37 Constructs | Kinase chaperone studies | Specific analysis of kinase folding and maturation pathways |
Implications for Therapeutic Development
The evolutionary conservation of chaperone families makes them attractive therapeutic targets. The structured organization into core and variable components across tissues provides opportunities for tissue-specific therapeutic interventions [3]. Understanding the precise mechanisms of Hsp70 and Hsp60 in protein folding offers potential for treating conformational diseases like Alzheimer's, Parkinson's, Huntington's, and ALS [5].
Current therapeutic strategies targeting chaperones have evolved through four developmental stages: (1) pan-isoform inhibitors (1990s), (2) isoform-selective inhibitors (2000s), (3) protein-protein interaction disruptors (2010s), and (4) multi-specific molecules (2020s) [6]. These approaches leverage the extensive structural and mechanistic knowledge of chaperone systems to develop targeted therapeutics.
The Hsp70 and Hsp60 chaperone families exhibit remarkable evolutionary conservation while demonstrating adaptive expansion and specialization across species. Their genomic organization reflects a balance between conserved core functions and tissue-specific adaptations essential for maintaining proteostasis. Integrated experimental approaches combining proteomics, genomics, and structural biology continue to unravel the complexity of chaperone networks. This knowledge provides the foundation for developing novel therapeutic strategies targeting the proteostasis network in human disease, particularly in cancer, neurodegenerative disorders, and aging-related conditions.
Molecular chaperones are fundamental components of the cellular proteostasis network, with Hsp70 and Hsp60 chaperones representing two paradigmatic systems that utilize distinct mechanistic strategies. Hsp70 employs a bidirectional allosteric mechanism between its nucleotide-binding domain (NBD) and substrate-binding domain (SBD) to interact with client proteins, whereas Hsp60 forms complex double-ring oligomeric structures that provide a protected folding chamber. This whitepaper provides an in-depth technical analysis of their domain architectures, allosteric regulation, and oligomerization dynamics, synthesizing recent structural and biochemical advances. The information is presented to equip researchers and drug development professionals with refined mechanistic understanding and methodological approaches for investigating these critical chaperone systems, with implications for therapeutic intervention in protein aggregation diseases.
Protein homeostasis, or proteostasis, relies on an integrated network of molecular chaperones that prevent aggregation, facilitate folding, and mediate remodeling of client proteins. The Hsp70 and Hsp60 families represent two essential, yet mechanistically distinct, chaperone systems. Hsp70 functions as a central hub in proteostasis through transient binding to short hydrophobic client segments, with its activity governed by an allosteric interplay between its NBD and SBD [7] [8]. In contrast, Hsp60 forms elaborate double-ring complexes that encapsulate clients within an isolated folding chamber, providing a protected environment for folding to occur [9] [10]. While both systems are ATP-dependent, their architectural principles and operational mechanisms differ significantly. Understanding their distinct domain organizations and functional regulations provides critical insights for developing targeted therapeutic strategies against proteostasis-related pathologies, including neurodegenerative diseases and cancer.
Hsp70: NBD-SBD Allosteric Communication
Domain Organization and Structural Basis
The Hsp70 chaperone consists of two principal domains: an N-terminal nucleotide-binding domain (NBD) of approximately 45 kDa and a C-terminal substrate-binding domain (SBD) of about 25 kDa [7] [11]. The NBD adopts an actin-like fold composed of four subdomains (IA, IB, IIA, IIB) arranged in two lobes that form a deep cleft for nucleotide binding [7]. The SBD is further divided into a β-sandwich subdomain (SBDβ) containing the substrate-binding groove and an α-helical lid subdomain (SBDα) [7] [8]. These domains are connected by a conserved, hydrophobic interdomain linker that serves as a critical structural element for allosteric coupling [7].
Table 1: Key Structural Elements in Hsp70 Allostery
| Structural Element | Domain Location | Functional Role | Conserved Residues |
|---|---|---|---|
| Interdomain linker | Connects NBD & SBD | Transmits allosteric signals | VLLL (in E. coli DnaK) |
| Nucleotide-binding cleft | NBD | Binds ATP/ADP; regulates conformation | Present in all four subdomains |
| Substrate-binding groove | SBDβ | Binds hydrophobic client sequences | Loops L1,2, L3,4, L4,5, L5,6 |
| α-helical lid | SBDα | Controls substrate access | Helices αA-αE |
| Hydrogen bond network | NBD-SBD interface | Mediates interdomain communication | R151, K155, R167, D481 (DnaK) |
The Allosteric Mechanism
Hsp70 function is governed by a bidirectional allosteric mechanism that couples nucleotide status with substrate-binding affinity:
ATP-bound state: ATP binding to the NBD induces substantial conformational changes, including subdomain re-orientations and rotation of lobe II [7]. This facilitates docking of both SBD subdomains onto the NBD, resulting in an open conformation where the α-lid detaches from the βSBD [7] [11]. This state exhibits low substrate affinity but fast association and dissociation rates, enabling rapid substrate binding and release [8].
ADP-bound state: ATP hydrolysis and ADP binding promote domain undocking, where the NBD and SBD behave as independent dynamic units [7]. The SBD adopts a closed conformation with the α-lid packed against the βSBD, creating a stable interface that results in high substrate affinity and slow substrate exchange rates [7] [11].
Allosteric coupling: The system operates as an energetic "tug-of-war" between two orthogonal interfaces: the βSBD-α-lid interface (favored in ADP-state) and the NBD-SBD interface (favored in ATP-state) [7]. Key residues, including D481 in E. coli DnaK, form a hydrogen bond network that mediates interdomain communication [11]. Substrate binding to the SBD allosterically stimulates ATP hydrolysis at the NBD, completing the bidirectional communication cycle [11] [8].
Figure 1: Hsp70 Allosteric Cycle. ATP binding induces a low-affinity, open state; hydrolysis yields a high-affinity, closed state; nucleotide exchange resets the cycle. Substrate binding stimulates ATP hydrolysis, enabling allosteric control.
Key Experimental Approaches and Reagents
Investigating Hsp70 allostery requires specialized methodologies and reagents:
Table 2: Research Reagent Solutions for Hsp70 Allostery Studies
| Reagent / Method | Specification / Function | Experimental Application |
|---|---|---|
| DnaK (E. coli Hsp70) | Model Hsp70 for mechanistic studies | Structural and biochemical analyses [7] [11] |
| TNP-ATP / MANT-nucleotides | Fluorescent nucleotide analogs | Monitoring nucleotide binding and kinetics [12] |
| Tryptophan fluorescence (W102) | Intrinsic fluorescence reporter | Detecting ATP-induced conformational changes [11] |
| Allosteric mutants (D481A/K) | Disrupt specific interdomain contacts | Probing allosteric communication pathways [11] |
| Single-turnover ATPase assays | Measures hydrolysis without exchange limitation | Assessing basal and stimulated ATPase rates [11] |
| ITC (Isothermal Titration Calorimetry) | Direct measurement of binding thermodynamics | Determining nucleotide affinity and stoichiometry [12] |
| Proteolysis sensitivity assays | Monitors domain docking/undocking | Probing conformational states under different conditions [12] |
Experimental Protocol: Analyzing Hsp70 Allosteric Mutants
- Protein Purification: Express and purify wild-type and mutant Hsp70 (e.g., D481A/K) using affinity and size-exclusion chromatography [11].
- Conformational Analysis: Monitor tryptophan fluorescence (W102 in DnaK) with excitation at 295 nm; ATP-induced blueshift indicates successful interdomain communication [11].
- Substrate Binding Kinetics: Use FRET-labeled peptides to measure substrate dissociation rates in presence of ATP versus ADP [11].
- ATPase Activity: Employ quenched-flow instruments for single-turnover ATP hydrolysis measurements; assess stimulation by J-domain proteins and substrates [11].
- Structural Validation: Utilize X-ray crystallography or cryo-EM to determine mutant structures; NMR for analyzing dynamics [7] [11].
Hsp60: Double-Ring Oligomerization and Function
Domain Architecture and Assembly
Mitochondrial Hsp60 (mtHsp60), a Group I chaperonin, shares structural homology with bacterial GroEL but exhibits distinct assembly properties. Each Hsp60 monomer consists of three domains: an equatorial domain that contains the ATP-binding site and mediates ring formation, an apical domain that binds substrates and the co-chaperonin Hsp10, and an intermediate domain that connects them and provides flexibility [9] [13]. Unlike GroEL, which forms stable tetradecamers, mtHsp60 exhibits dynamic oligomerization that depends on nucleotide presence [9] [10].
Table 3: Hsp60 Oligomeric States and Properties
| Oligomeric State | Structural Features | Functional Capacity | Stability |
|---|---|---|---|
| Monomer | Single subunit; disordered interaction interfaces | No ATPase activity; cannot assist folding [10] [13] | Low; prone to aggregation |
| Heptamer (Single-ring) | Seven subunits arranged in a ring; open at one end | Active folding with Hsp10; functional but less efficient than double-ring [9] [10] | Moderate; requires nucleotide for stability |
| Tetradecamer (Double-ring) | Two back-to-back heptameric rings; symmetrical structure | High-efficiency folding machine; encapsulates substrates [9] | High; stabilized by ATP and Hsp10 |
Oligomerization Mechanism and Functional Cycle
Hsp60 assembly and function are governed by nucleotide-dependent conformational changes:
Nucleotide-driven assembly: ATP binding promotes heptamer formation and subsequent double-ring assembly, unlike GroEL which forms stable tetradecamers without nucleotide [9]. The equatorial domain contains critical ATP-binding motifs (e.g., 85DGTTT89, G414, D494 in human mtHsp60) that mediate inter-subunit interactions [13].
Functional states: Hsp60 exists in equilibrium between single-ring "half-football" complexes (Hsp607-Hsp107) and double-ring "football" complexes (Hsp6014-(Hsp107)2) [9]. Both assemblies are functionally active in protein folding, with the double-ring form representing the ground state [9].
Lack of negative cooperativity: Unlike GroEL, Hsp60 does not display negative ATP-binding inter-ring cooperativity, allowing simultaneous ATP binding to both rings [9]. This fundamental mechanistic difference impacts the folding cycle and potential regulatory mechanisms.
Folding cycle: Client proteins bind to hydrophobic residues in the apical domain; ATP binding induces conformational changes that promote Hsp10 binding and encapsulation of the client within the folding chamber; ATP hydrolysis triggers release of the folded client [9] [14].
Figure 2: Hsp60 Oligomerization and Functional Cycle. ATP binding drives assembly from monomers to single rings, then to double rings; Hsp10 binding forms active football complexes; hydrolysis triggers disassembly.
Key Experimental Approaches for Hsp60 Oligomerization
Studying Hsp60 oligomerization requires techniques that capture dynamic assembly states:
Table 4: Research Reagent Solutions for Hsp60 Oligomerization Studies
| Reagent / Method | Specification / Function | Experimental Application |
|---|---|---|
| SEC-MALS | Size-exclusion chromatography with multi-angle light scattering | Determining oligomeric states and molecular weights in solution [9] |
| ADP:BeF3 | ATP ground-state mimic | Trapping and stabilizing football complexes for structural studies [9] |
| Cryo-EM single particle analysis | High-resolution structure determination of complexes | Visualizing football and half-football complexes at near-atomic resolution [9] |
| BS3 crosslinker | Chemical crosslinking with amine-reactive NHS esters | Stabilizing and identifying transient oligomeric states [13] |
| Temperature-controlled purification | Manipulating assembly stability | Isolating specific oligomeric forms (dimers at 4°C, heptamers at 25°C) [13] |
| Negative-stain TEM | Rapid structural assessment | Confirming ring-shaped structures and oligomeric states [13] |
| Obligate single-ring mutants | Engineered variants deficient in double-ring formation | Probing functional significance of different oligomeric states [9] |
Experimental Protocol: Characterizing Hsp60 Oligomeric States
- Temperature-Modulated Purification: Purify Hsp60 at 4°C to isolate dimeric forms and at 25°C to obtain heptameric/tetradecameric complexes [13].
- Complex Stabilization: Incubate Hsp60 with ATP and BeF3 to stabilize football complexes for structural studies [9].
- Oligomeric State Analysis: Use SEC-MALS to determine molecular weights and distributions of different oligomeric species in solution [9] [13].
- ATPase Activity Assays: Measure ATP hydrolysis kinetics across different oligomeric states; dimeric Hsp60 shows deficient ATPase activity [13].
- Structural Characterization: Apply single-particle cryo-EM to determine structures of football and half-football complexes at 3.0-3.8 Å resolution [9].
- Functional Complementation: Test obligate single-ring and double-ring mutants for in vivo function using bacterial complementation assays [9].
Comparative Analysis and Research Implications
Mechanistic Comparison of Hsp70 and Hsp60
Despite sharing fundamental roles in proteostasis, Hsp70 and Hsp60 employ strikingly different mechanistic strategies:
Table 5: Comparative Analysis of Hsp70 and Hsp60 Chaperone Systems
| Characteristic | Hsp70 System | Hsp60 System |
|---|---|---|
| Fundamental Mechanism | Allosteric regulation between NBD and SBD | Oligomeric assembly into folding chambers |
| Domain Architecture | Two-domain monomer (NBD + SBD) with flexible linker | Three-domain monomer (apical, intermediate, equatorial) |
| Functional Oligomerization | Primarily monomeric; cooperates with J-proteins and NEFs | Essential oligomerization (heptameric rings and tetradecamers) |
| Nucleotide Regulation | ATP/ADP cycling controls substrate affinity | ATP binding drives assembly and folding cycle |
| Substrate Recognition | Short hydrophobic stretches in extended conformations | Broader structural elements in folding intermediates |
| Co-chaperone Requirements | J-domain proteins and nucleotide exchange factors | Hsp10 (co-chaperonin) lid structure |
| Folding Environment | Transient binding without encapsulation | Sequestration in protected central cavity |
| Allosteric Properties | Bidirectional communication between domains | Inter-ring communication without negative cooperativity |
Research Applications and Therapeutic Implications
The distinct mechanisms of Hsp70 and Hsp60 present unique research and therapeutic opportunities:
Hsp70 as a therapeutic target: The allosteric regulation of Hsp70 makes it an attractive target for diseases of proteostasis. Small molecules that modulate the allosteric interface could potentially correct dysregulated chaperone function in neurodegenerative diseases and cancer [15] [16].
Hsp60 in disease contexts: The dynamic oligomerization of Hsp60 and its presence in extramitochondrial locations under pathological conditions suggests context-specific functions [10]. Different oligomeric states may play distinct roles in cancer progression and neurodegeneration, offering potential for selective therapeutic targeting [10].
Experimental design considerations: Research on these chaperones requires careful consideration of their dynamic nature. Hsp70 studies must account for nucleotide state and co-chaperone interactions, while Hsp60 research requires controls for oligomeric equilibrium and stabilization conditions [11] [13].
Technical advancements: Recent cryo-EM breakthroughs have enabled visualization of transient Hsp60 football complexes, while sophisticated NMR and single-molecule techniques have revealed Hsp70 allosteric dynamics at unprecedented resolution [7] [9].
The domain architecture of Hsp70, characterized by sophisticated NBD-SBD allostery, and the double-ring oligomerization of Hsp60 represent two elegant evolutionary solutions to the fundamental challenge of protein folding in the cellular environment. While Hsp70 operates through a dynamic, allosterically-regulated clamp mechanism ideal for binding transiently exposed hydrophobic segments, Hsp60 forms structured folding chambers that encapsulate clients entirely. Both systems exemplify the intricate relationship between protein structure and function in maintaining proteostasis. Their continued investigation not only deepens our understanding of basic cellular processes but also opens promising avenues for therapeutic intervention in the growing spectrum of protein conformation diseases. Future research directions include elucidating the full structural dynamics of these chaperones in their native environments, developing more specific chemical modulators, and exploring the therapeutic potential of manipulating their oligomeric states and allosteric networks.
Within the cellular machinery, molecular chaperones are essential guardians of protein homeostasis (proteostasis), ensuring the proper folding, assembly, and localization of proteins and mitigating the damage caused by proteotoxic stress [17] [18]. The 70 kDa and 60 kDa heat shock proteins, Hsp70 and Hsp60, are two central pillars of this chaperone network. Despite sharing a common goal of maintaining proteostasis, they employ distinct, sophisticated mechanisms centered on ATP-driven conformational switches to manage their client proteins [17]. This review delineates the core reaction cycles of Hsp70 and Hsp60, highlighting how ATP binding and hydrolysis power conformational changes that enable these chaperones to bind, fold, and release a diverse array of client substrates. A deep understanding of these mechanisms is paramount for developing novel therapeutic strategies against cancers, neurodegenerative diseases, and other conditions linked to chaperone malfunction [17] [6].
The Hsp70 Chaperone Cycle
Domain Architecture and Allosteric Regulation
Hsp70 chaperones are multi-domain proteins conserved from bacteria to humans. They consist of two principal structured domains [19] [18]:
- A 45 kDa N-terminal Nucleotide-Binding Domain (NBD)
- A 25 kDa C-terminal Substrate-Binding Domain (SBD) The SBD is further subdivided into a β-sandwich base (SBDβ) that forms the client binding pocket and an α-helical lid (SBDα). These domains are connected by a conserved, flexible linker that is critical for allosteric communication [18]. The NBD binds ATP and undergoes significant conformational changes during its hydrolysis. The SBD engages client proteins, preferring short stretches of hydrophobic amino acids flanked by basic residues [19] [20].
Table 1: Key Structural Domains of Hsp70 and Their Functions
| Domain | Molecular Weight | Primary Function | Key Structural Features |
|---|---|---|---|
| N-terminal Nucleotide-Binding Domain (NBD) | ~45 kDa | ATP binding and hydrolysis | Four subdomains (IA, IB, IIA, IIB) forming a cleft for nucleotide binding [19] [21] |
| C-terminal Substrate-Binding Domain (SBD) | ~25 kDa | Client protein recognition and binding | SBDβ (base): hydrophobic binding groove; SBDα (lid): regulates client access [19] [18] |
| Flexible Linker | Variable | Allosteric communication | Connects NBD and SBD; conformation changes with nucleotide state [18] |
The ATP-Driven Conformational Switch
The functional cycle of Hsp70 is governed by an allosteric mechanism that couples nucleotide state in the NBD to client affinity in the SBD [18].
- ATP-Bound State (Open Conformation): When ATP is bound in the NBD, the Hsp70 complex adopts an open conformation. The SBDα lid is retracted, and the SBDβ exhibits low affinity for client proteins. This state allows for rapid binding and release of client substrates [18].
- ATP Hydrolysis and Client Trapping: The ATPase activity of Hsp70, while intrinsically low, is dramatically stimulated by J-protein co-chaperones (Hsp40). Hsp40 delivers client proteins to the SBD and interacts with the Hsp70 NBD to trigger ATP hydrolysis [19] [18]. The energy from hydrolysis is transduced via the linker, causing the SBDα lid to close over the client protein, trapping it in the SBD with high affinity [18].
- ADP-Bound State (Closed Conformation): In the ADP-bound state, the chaperone remains in a closed conformation, stably associated with the client. This provides a protected environment, preventing the client from aggregating [17].
- Nucleotide Exchange and Client Release: The release of ADP is rate-limiting and is accelerated by Nucleotide Exchange Factors (NEFs) such as GrpE in bacteria or Bag-1 and Hsp110 in eukaryotes. NEFs pry the NBD open, facilitating the exchange of ADP for ATP. Once ATP binds, the cycle resets to the open conformation, releasing the folded or now-competent client protein [19] [21] [18].
This cycle enables Hsp70 to function as a "multiple socket," providing a platform where client proteins can be handed off to other chaperones or degradation systems based on the specific co-chaperones present [18].
Mechanism of Client Recognition
A fundamental characteristic of Hsp70 is its mode of client recognition. Research using solution NMR spectroscopy has established that both bacterial and human Hsp70 interact with clients via a conformational selection (CS) mechanism [20]. Hsp70 does not actively unfold its clients; instead, it selectively binds to pre-existing unfolded or partially unfolded conformations from a dynamic ensemble of interconverting client structures. This ensures that Hsp70 primarily engages non-native proteins while leaving natively folded proteins undisturbed [20].
Diagram 1: The Hsp70 ATP-driven chaperone cycle.
The Hsp60 Chaperonin Cycle
Structure and Classification
Hsp60 chaperonins, also known as Cpn60, form large, multi-subunit complexes that provide an encapsulated folding chamber for client proteins. They are classified into two groups [1] [22]:
- Group I: Found in eubacteria (GroEL), mitochondria (mtHSP60), and chloroplasts. They form a double-ring structure of 7 subunits per ring and require a dedicated co-chaperone lid (GroES in bacteria, HSP10 in eukaryotes) to function [1].
- Group II: Found in archaea and the eukaryotic cytosol (TRiC/CCT). They form double rings of 8 or 9 subunits per ring and have a built-in lid structure, making them independent of a GroES-like co-chaperone [1] [22].
The prototypical Group I chaperonin, GroEL, is a tetradecamer arranged in two back-to-back heptameric rings. Each subunit comprises three domains [1]:
- Equatorial Domain: Contains the ATPase activity and provides the main inter-ring and intra-ring contacts.
- Apical Domain: Forms the opening of the folding chamber and contains hydrophobic residues for client binding.
- Intermediate Domain: Connects the equatorial and apical domains and acts as a hinge during conformational changes.
Table 2: Comparison of Group I and Group II Chaperonins
| Feature | Group I Chaperonins (e.g., GroEL, mtHSP60) | Group II Chaperonins (e.g., TRiC/CCT) |
|---|---|---|
| Organisms | Eubacteria, Mitochondria, Chloroplasts | Archaea, Eukaryotic Cytosol |
| Ring Structure | Two heptameric rings (7+7) | Two octameric or nonameric rings (8+8/9+9) |
| Lid Mechanism | Requires separate GroES/HSP10 co-chaperone lid | Built-in lid formed by apical domain extensions |
| Folding Chamber | Encapsulates client in an isolated environment | Encapsulates client in an isolated environment |
The Folding Cycle and Coordinated Conformational Changes
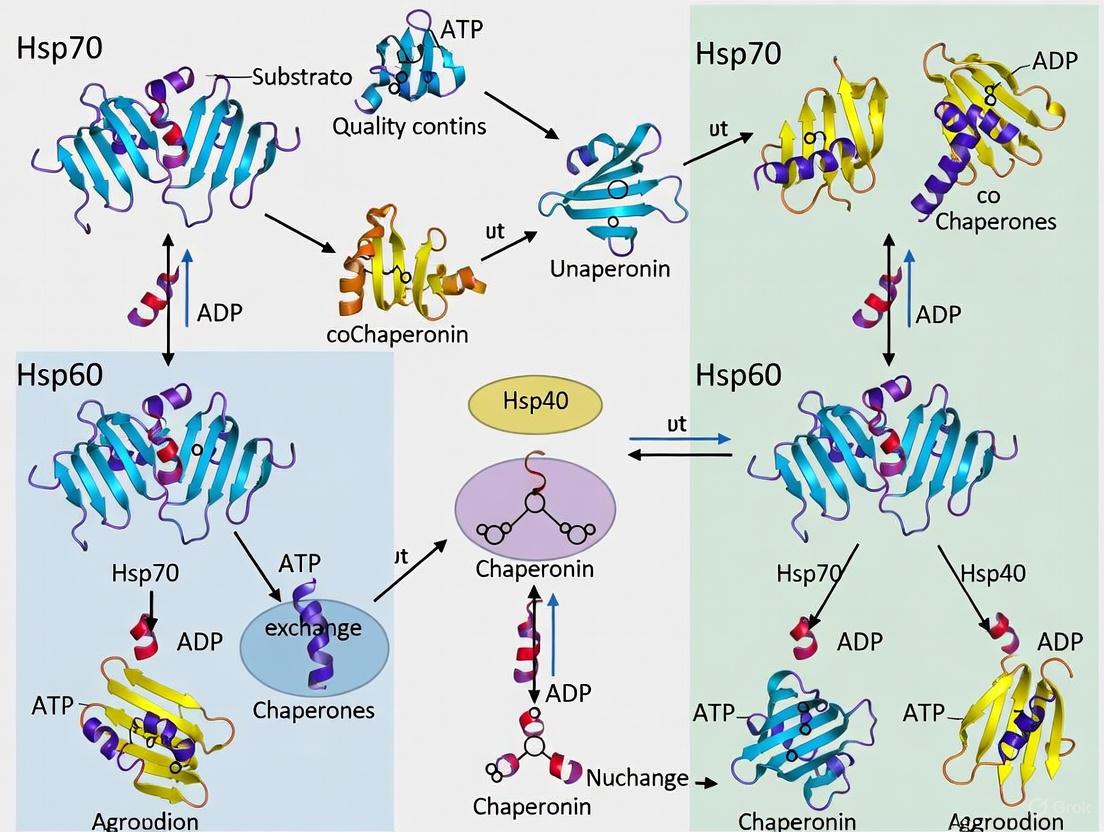
The GroEL/GroES reaction cycle is a finely tuned process driven by ATP binding and hydrolysis, characterized by negative cooperativity between the two rings: when one ring is in a high-affinity state for ATP, the other is in a low-affinity state [1] [22].
Diagram 2: The GroEL/GroES (Hsp60/Hsp10) folding cycle.
The cycle proceeds as follows [1]:
- Client Binding: An unfolded client protein binds to the hydrophobic apical domains of one ring (the cis ring) of GroEL, which is in a nucleotide-free or ADP-bound state.
- Encapsulation: The binding of ATP and the co-chaperone GroES to the same cis ring triggers dramatic, concerted conformational changes. The apical domains undergo an upward and outward rotation, displacing the client protein into the now-enlarged and hydrophilic folding chamber. The chamber becomes encapsulated, isolating the client from the bulk cytosol.
- Folding: The client protein has a defined timeframe (the timescale of ATP hydrolysis, ~10-15 seconds) to fold within this protected Anfinsen cage.
- Release: The binding of ATP and a new client protein to the opposite trans ring acts as a allosteric trigger that discharges GroES, ADP, and the folded (or folding-competent) client from the cis ring. If the client has not reached its native state, it may rebind for another round of folding.
This alternating cycle of the two rings ensures continuous client processing [1] [22].
Experimental Analysis of Chaperone Mechanisms
Key Methodologies and Reagents
Understanding the conformational dynamics and client interactions of Hsp70 and Hsp60 relies on a suite of sophisticated biophysical and biochemical techniques.
Table 3: Key Experimental Protocols for Studying Chaperone Mechanisms
| Methodology | Application & Rationale | Key Experimental Insights |
|---|---|---|
| Site-Directed Mutagenesis | Probing functional residues (e.g., ATPase site, client binding pocket). An R261A mutation in Hsp70 NBD was shown to alter ATP kinetics and fluorescence quenching [21]. | Validated computational predictions of allosteric communication pathways and identified critical residues for function [21]. |
| All-Atom Molecular Dynamics (MD) Simulations | Modeling atomic-level conformational dynamics and allosteric transitions. Simulations of Hsp70 NBD revealed a more closed ATP-bound state than seen in crystal structures [21]. | Predicted novel conformational states and provided atomic-level mechanisms for allostery and conformational selection [21] [20]. |
| Solution NMR Spectroscopy | Characterizing structure, dynamics, and binding events under native conditions. Used to quantify conformational selection in Hsp70-client interactions [20]. | Established that Hsp70 selects unfolded client conformations from a pre-existing ensemble, and defined the structural state of clients when bound [20]. |
| Fluorescence Spectroscopy | Monitoring conformational changes in real-time. Intrinsic tryptophan fluorescence (e.g., Trp90 in Hsp70) reports on nucleotide-dependent domain rearrangements [21]. | Provided kinetic data on ATP-induced conformational changes and validated allosteric models derived from structures and simulations [21]. |
| X-ray Crystallography & Cryo-EM | Determining high-resolution structures of chaperones and their complexes. Revealed structures of GroEL/GroES and various Hsp70 conformational states [1] [6]. | Provided the foundational structural frameworks for understanding domain organization and large-scale quaternary changes [1] [6]. |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Chaperone Studies
| Reagent / Tool | Function in Research | Example Application |
|---|---|---|
| Recombinant Chaperones (Hsp70, Hsp60, GroEL/ES) | Purified proteins for in vitro folding assays, structural studies, and biochemical characterization. | Core component for ATPase assays, client refolding experiments, and structural biology [1] [20]. |
| Co-chaperone Proteins (Hsp40, GrpE, Bag-1, Hsp110) | Regulate the ATPase cycle of Hsp70. Essential for reconstituting functional cycles in vitro. | Used to study stimulation of ATP hydrolysis (Hsp40) and nucleotide exchange (GrpE, Bag-1) [19] [18]. |
| Site-Directed Mutants (e.g., R261A Hsp70) | To dissect the functional contribution of specific amino acid residues. | The R261A Hsp70 mutant helped confirm the role of Arg261 in ATP-induced conformational changes and fluorescence quenching [21]. |
| Isotope-labeled Substrates (e.g., 15N, 13C-labeled clients) | Enables high-resolution NMR studies of chaperone-client interactions and client conformations. | Critical for determining that Hsp70 binds clients via conformational selection [20]. |
| Fluorescent Nucleotide Analogs (e.g., mant-ATP/ADP) | Report on nucleotide binding and dissociation kinetics. | Used in stopped-flow experiments to measure nucleotide affinity and the effects of NEFs and clients [21]. |
Hsp70 and Hsp60 exemplify nature's sophisticated solutions to the complex problem of protein homeostasis. While both are ATP-dependent molecular machines, their mechanisms are distinct. Hsp70 operates as a versatile "triage" chaperone, using a allosteric two-domain switch to selectively bind and release unfolded clients in a process governed by conformational selection and tightly regulated by co-chaperones [20] [18]. In contrast, Hsp60 functions as a specialized folding cage, utilizing a complex, cooperative two-ring system to physically encapsulate a single client protein, providing it with a protected environment to reach its native conformation [1] [22]. The continued elucidation of these ATP-driven cycles, through integrative structural biology, biophysics, and computation, not only deepens our fundamental understanding of cellular physiology but also paves the way for targeting these essential chaperones in human disease [17] [6].
Within the complex cellular environment, the maintenance of protein homeostasis (proteostasis) is a fundamental process, with molecular chaperones serving as the primary machinery for ensuring proper protein folding, preventing aggregation, and mediating cellular stress responses [1] [23]. The 70-kDa heat shock protein (Hsp70) and chaperonin (Hsp60) families represent two central pillars of this proteostasis network. While these chaperones form the core folding machinery, their functional specificity and regulation are critically dependent on dynamic interactions with co-chaperones [24] [25]. This orchestration is achieved through specialized co-chaperone networks: J-domain proteins (JDPs) and nucleotide exchange factors (NEFs) for the Hsp70 system, and Hsp10 for the Hsp60 system. These co-chaperones transform generic chaperone scaffolds into precise, regulated machines capable of handling a diverse array of client proteins and complex cellular challenges [26] [27]. Understanding the mechanisms of these cooperative systems is not only fundamental to cell biology but also provides crucial insights for developing therapeutic interventions for protein misfolding diseases, including neurodegeneration and cancer [1] [25].
The Hsp70 Chaperone System
The Hsp70 system is a versatile chaperone machine involved in a wide range of cellular processes, including de novo protein folding, prevention of protein aggregation, membrane translocation, and disassembly of protein complexes [23]. Its function is governed by an allosteric ATPase cycle that regulates its affinity for client proteins.
Core Mechanism of the Hsp70 Chaperone Cycle
Hsp70 consists of two primary domains: an N-terminal nucleotide-binding domain (NBD) that hydrolyzes ATP, and a C-terminal substrate-binding domain (SBD) that interacts with client proteins [25] [23]. The chaperone alternates between two conformational states:
- ATP-bound state: The SBD exhibits fast substrate association and dissociation rates, characterized by an open lid structure.
- ADP-bound state: The SBD has slow substrate kinetics, with a closed lid that tightly traps client proteins.
The intrinsic ATPase activity of Hsp70 is low, and the switching between these states requires precise regulation by co-chaperones to achieve efficient client protein folding and processing [25] [23].
J-domain Proteins (JDPs): Specificity Factors for Hsp70
J-domain proteins (JDPs), also known as Hsp40s, constitute the largest family of Hsp70 co-chaperones and play a determinative role in specifying Hsp70 functions [24] [26].
Table 1: Classification of J-domain Proteins (JDPs)
| Class | Domain Architecture | Representative Members | Key Features |
|---|---|---|---|
| Class A | J-domain → G/F region → Zinc-finger domain → C-terminal domain (CTD) | E. coli DnaJ, Yeast Ydj1 | Most similar to bacterial DnaJ; contain zinc finger motifs |
| Class B | J-domain → G/F region → Domain without zinc finger | Human DNAJB1, Yeast Sis1 | Lack zinc-binding motifs; may have other substrate interaction domains |
| Class C | J-domain with varied other domains | Diverse species-specific JDPs | Largest class; diverse domain architectures beyond typical JDP structure |
JDPs share a defining ~70 amino acid J-domain that contains a conserved His-Pro-Asp (HPD) motif essential for stimulating Hsp70's ATPase activity [26]. The J-domain interacts with the ATP-bound state of Hsp70, contacting lobe IIA of the NBD, the inter-domain linker, and the β-sandwich of the SBD [26]. This multi-site interaction couples substrate binding to ATP hydrolysis, enabling the synergistic action of substrates and J-domains in stimulating the Hsp70 ATPase cycle [26].
Beyond their J-domains, these co-chaperones possess diverse additional domains that drive functional specificity by delivering particular substrates or recruiting Hsp70 to specific cellular locations [24] [26]. In multicellular organisms, JDP expression shows tissue and cell-type heterogeneity, allowing specialized proteostasis networks tailored to distinct cellular environments [26].
Nucleotide Exchange Factors (NEFs): Completing the Hsp70 Cycle
Following ATP hydrolysis and client protein binding, the release of ADP from Hsp70 becomes rate-limiting for the chaperone cycle. Nucleotide Exchange Factors (NEFs) catalyze this critical step, facilitating the transition from the ADP-bound to ATP-bound state and subsequent client release [28] [25].
Table 2: Nucleotide Exchange Factor (NEF) Families in Eukaryotes
| NEF Family | Structure | Representative Members | Mechanistic Role |
|---|---|---|---|
| Hsp110 (Hsp70-related) | NBD, β-sandwich, 3-helix bundle | Mammalian Hsp110, Yeast Sse1p/Sse2p | Binds Hsp70 NBD, induces conformational change for ADP release |
| BAG Domain | BAG domain | BAG-1, BAG-3 | Interacts with Hsp70 NBD, accelerates nucleotide exchange |
| HspBP1 | Armadillo repeats | HspBP1, Fes1p | Binds Hsp70 NBD, disrupts nucleotide binding site |
| GrpE | Dimeric structure | Mitochondrial GrpE | Bacterial homolog; functions as homodimer |
The Hsp110 family represents a specialized subclass of NEFs that are themselves homologs of Hsp70 but have evolved to primarily function as NEFs rather than canonical chaperones [28]. Structural studies of the yeast Hsp110 Sse1 in complex with the Hsp70 NBD reveal that Hsp110 embraces the Hsp70 NBD, inducing structural changes that open the nucleotide-binding cleft and facilitate ADP release [29]. This NEF activity is essential for cellular viability, as demonstrated by the synthetic lethality observed in yeast lacking both SSE1 and SSE2 genes [28].
The functional importance of this regulation is evidenced by experimental findings that equimolar amounts of Sse1p increase the rate of nucleotide exchange on the yeast Hsp70 Ssa1p by approximately 12-fold, significantly outperforming other NEFs like Fes1p [28]. This efficient nucleotide exchange directly enhances Hsp70-mediated refolding of denatured proteins, as demonstrated with thermally denatured firefly luciferase [28].
The Hsp60 Chaperonin System
The Hsp60 family, also known as chaperonins, represents a distinct class of chaperones that form large, barrel-shaped complexes that provide an isolated environment for protein folding [1] [27].
Structural Organization and Classification
Chaperonins are classified into two main groups based on their structure and occurrence:
Table 3: Classification of Chaperonin Systems
| Group | Representative Complexes | Structure | Co-chaperone Requirement | Cellular Localization |
|---|---|---|---|---|
| Group I | GroEL (bacteria), HSP60/HSP10 (mitochondria) | Double 7-mer ring | Hsp10 (GroES) co-chaperone essential | Bacteria, mitochondria, chloroplasts |
| Group II | TRiC/CCT (eukaryotic cytosol), Thermosome (archaea) | Double 8-9-mer ring | Built-in lid; prefoldin co-factor | Eukaryotic cytosol, archaea |
Group I chaperonins, such as the well-characterized GroEL/GroES system in E. coli and the homologous HSP60/HSP10 system in mammalian mitochondria, form symmetrical double-ring complexes with seven subunits per ring [1] [27]. Each subunit consists of three domains: equatorial (mediating ring-ring interactions and ATP binding), intermediate (acting as a hinge), and apical (containing the substrate-binding sites) [27].
Hsp10 Cooperation in the Chaperonin Folding Cycle
Hsp10 (known as GroES in bacteria) functions as a essential co-chaperone for Group I chaperonins, forming a dome-like structure that seals the folding chamber [1] [27]. The cooperative mechanism follows a well-defined sequence:
- Client protein binding: Unfolded substrate proteins bind to hydrophobic residues in the apical domains of the open GroEL (Hsp60) ring.
- Hsp10 binding: ATP and Hsp10 bind cooperatively to the same ring, causing dramatic conformational changes that elevate and twist the apical domains.
- Folding encapsulation: The Hsp10 "lid" seals the chamber, creating an isolated environment that shields the client protein from the crowded cellular milieu.
- Folding and release: ATP hydrolysis in the cis-ring and ATP binding to the trans-ring trigger Hsp10 and substrate release, typically after a complete catalytic cycle of approximately 10-15 seconds.
This mechanism allows proteins up to ~60 kDa to fold within the protected central cavity, which provides essential isolation from potential aggregation partners in the cellular environment [27]. The encapsulation event also induces a conformational shift from hydrophobic to hydrophilic surfaces in the folding chamber, promoting proper protein folding rather than aggregation [1].
Experimental Approaches for Studying Co-chaperone Networks
Key Methodologies and Reagents
Advanced experimental techniques have been essential for elucidating the mechanisms of co-chaperone networks.
Table 4: Key Research Reagents and Experimental Approaches
| Research Tool | Experimental Application | Key Findings Enabled |
|---|---|---|
| Fluorescent MABA-ADP | Stopped-flow fluorimetry to measure nucleotide exchange rates | Quantitative analysis of NEF activity; Sse1p accelerates Ssa1p nucleotide release 12-fold [28] |
| Recombinant Hsp70/Hsp40/NEF systems | In vitro refolding assays with denatured substrates (e.g., firefly luciferase) | Demonstration that optimal Sse1p concentrations boost Hsp70-mediated refolding yield and rate [28] |
| X-ray crystallography of Sse1p-Hsp70 complexes | Structural analysis of co-chaperone:chaperone interactions | Revealed how Hsp110 embraces Hsp70 NBD to induce ADP release [29] |
| Yeast genetics (SSE1/SSE2 deletion strains) | In vivo analysis of co-chaperone function | Established essential role of Hsp110 NEF activity; synthetic lethality of double deletions [28] |
| Small molecule inhibitors (MKT-077, JG-98) | Modulating Hsp70-co-chaperone interactions | Identification of allosteric sites that disrupt Hsp70-BAG interactions; potential therapeutic applications [25] |
Experimental Workflow Diagram
The following diagram illustrates a representative experimental approach for analyzing nucleotide exchange factor activity, a key methodology in co-chaperone research:
Diagram 1: Experimental workflow for measuring NEF activity using fluorescent MABA-ADP. The fluorescence of MABA-ADP decreases upon release from Hsp70, allowing quantification of nucleotide exchange rates [28].
Chaperone-Co-chaperone Functional Relationships
The functional integration between chaperones and their co-chaperones can be visualized as a coordinated network:
Diagram 2: Functional relationships in chaperone-co-chaperone networks. JDPs target substrates to Hsp70 and stimulate ATP hydrolysis, while NEFs reset the cycle by promoting ADP release. In the Hsp60 system, Hsp10 forms a sealed folding chamber for client encapsulation [28] [1] [26].
Implications for Disease and Therapeutic Development
The critical role of co-chaperone networks in proteostasis has profound implications for human disease, particularly in neurodegeneration and cancer [25] [30].
In neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, dysfunction of the Hsp70 co-chaperone system contributes to the accumulation of misfolded proteins [25]. For example, Hsp70 in complex with specific JDPs can disassemble tau fibrils but may generate seeding-competent oligomeric tau species in the process [25]. Similarly, mutations in synaptic JDPs like DNAJC5 (CSPα) and DNAJC6 (auxilin) cause adult neuronal ceroid lipofuscinosis and early-onset Parkinson's disease, respectively, highlighting the specialized functions of these co-chaperones in neuronal proteostasis [30].
Therapeutic strategies are increasingly targeting specific co-chaperone interactions rather than the general chaperone machinery to avoid detrimental side effects [25]. Small molecules like JG-98 that allosterically inhibit Hsp70-BAG interactions have shown promise in modulating chaperone function for therapeutic benefit, illustrating the potential of targeting co-chaperone networks for drug development [25].
Co-chaperone networks represent sophisticated regulatory systems that confer specificity, efficiency, and functional diversity to the core chaperone machinery. J-domain proteins and nucleotide exchange factors partner with Hsp70 to form a dynamic system capable of responding to diverse protein folding challenges, while Hsp10 cooperation with Hsp60 creates an essential protected environment for proper protein folding. Continued elucidation of these cooperative mechanisms will not only advance our fundamental understanding of cellular proteostasis but also provide novel therapeutic avenues for the growing number of diseases associated with protein misfolding and aggregation. The integrated view presented in this review highlights the complexity and elegance of these essential cellular systems and underscores their importance in both health and disease.
Within the cellular milieu, molecular chaperones are fundamental components of the protein homeostasis (proteostasis) network, responsible for recognizing, binding, and assisting in the folding of non-native proteins. Their function prevents the toxic consequences of protein aggregation, a hallmark of numerous age-related diseases [17] [31]. The Hsp70 and Hsp60 chaperone families are central to this proteostasis machinery, utilizing distinct but complementary mechanisms to manage substrate proteins. The core principle underlying substrate recognition across these chaperone families is the specific detection of hydrophobic amino acid patches that are exposed in non-native, misfolded, or unfolding polypeptides [32] [33]. These hydrophobic surfaces, which are typically buried within the core of correctly folded proteins, represent a danger of aberrant interactions that can lead to aggregation. By binding these patches, chaperones effectively shield them, thereby preventing aggregation and allowing folding to proceed correctly [33] [31]. This whitepaper provides an in-depth technical analysis of the mechanisms by which Hsp70 and Hsp60 chaperones recognize their substrates and prevent aggregation, framed within the context of modern proteostasis research for a scientific audience.
Hsp70 Chaperone System: Mechanism of Substrate Recognition
Structural Domains and Allosteric Regulation
The Hsp70 chaperone system, comprising Hsp70 (DnaK in bacteria) and its co-chaperones, operates as a central hub in proteostasis, assisting in a wide range of processes including de novo folding, refolding, and translocation [17] [33]. Hsp70 proteins share a conserved architecture consisting of two primary domains:
- N-terminal Nucleotide-Binding Domain (NTD): This domain exhibits ATPase activity.
- C-terminal Substrate-Binding Domain (SBD): This domain is responsible for binding client proteins.
These two domains are connected by a flexible linker [17]. The SBD is further subdivided into a β-sandwich subdomain (SBDβ) that houses the hydrophobic peptide-binding pocket and an α-helical lid (SBDα) [17]. The chaperone activity of Hsp70 is governed by an allosteric mechanism tied to its ATPase cycle. When the NTD is bound to ATP, the SBD adopts an open conformation characterized by low affinity for substrates and fast binding/dissociation kinetics. Hydrolysis of ATP to ADP triggers a conformational shift that closes the lid, resulting in a high-affinity state that stably traps the substrate [17] [33]. This cycle is critically regulated by co-chaperones; Hsp40 (DnaJ) proteins act as J-proteins that stimulate Hsp70's ATPase activity and often deliver initial substrates, while nucleotide exchange factors (e.g., GrpE in E. coli) promote the release of ADP, allowing a new ATP to bind and substrate release [17] [6].
Specificity for Hydrophobic Patches
Hsp70 chaperones recognize short, degenerate peptide stretches of approximately 7 amino acids within their substrate proteins [34]. The core recognition motif is hydrophobic in nature, typically enriched with branched-chain aliphatic amino acids like leucine and isoleucine [34] [33]. The SBDβ domain contains a specific pocket that accommodates these hydrophobic residues, while flanking regions of the substrate peptide may interact with more polar areas of the binding groove. This mechanism allows Hsp70 to bind to a vast repertoire of partially folded or unfolded proteins, as these hydrophobic segments are transiently exposed during folding or become permanently accessible in misfolded states [33]. The primary role of the Hsp70 system is often described as a "holdase" or "gatekeeper," minimizing aggregation of nascent chains and unfolded proteins by binding hydrophobic patches, thereby keeping them in a folding-competent state before they may be transferred to other chaperone systems like Hsp60 for active folding [33].
Hsp60 Chaperone System: Encapsulation for Folding
The GroEL-GroES Complex Architecture
In contrast to the more open binding mode of Hsp70, the Hsp60 chaperonin system, exemplified by the GroEL-GroES complex in bacteria, provides an encapsulated environment for protein folding. GroEL is a large, double-ring structure composed of 14 identical subunits arranged in two heptameric rings stacked back-to-back [17]. Each ring defines a central cavity that serves as the folding chamber. The co-chaperonin GroES is a single heptameric ring that acts as a lid for the GroEL complex [17] [33].
The internal surface of the GroEL cavity is predominantly hydrophilic, which provides an environment that discourages aggregation and promotes proper folding, in stark contrast to the hydrophobic binding surfaces used for initial substrate recognition [33]. The folding process is ATP-dependent. A partially folded substrate protein is captured by the hydrophobic apical domains of the open GroEL ring. ATP binding to the ring facilitates the binding of GroES, which triggers a massive conformational change: the apical domains rotate and elevate, sequestering the substrate inside the now hydrophilic, encapsulated chamber. This process promotes folding during a timescale set by the ATP hydrolysis cycle within the ring. Upon ATP hydrolysis in the encapsulated ring and subsequent ATP binding to the opposite ring, the lid is released, and the substrate, whether folded or not, is discharged [17] [33].
Recognition at the Hydrophobic Apical Domain
Substrate recognition by GroEL does not rely on a specific linear sequence motif but is mediated by flexible hydrophobic residues located in the apical domains of the GroEL subunits [33]. These hydrophobic patches, which include residues such as Val, Leu, and Ile, interact with complementary hydrophobic surfaces exposed on non-native substrate proteins. This interaction is relatively promiscuous, allowing GroEL to assist in the folding of a diverse set of cytosolic proteins, typically in the size range of 20-60 kDa [33]. The binding itself helps to prevent off-pathway aggregation interactions. The subsequent conformational change that occurs upon GroES binding serves a critical dual function: it physically separates the substrate from the bulk solvent and other folding intermediates, and it removes the substrate from the initial hydrophobic binding sites, shifting it into the hydrophilic folding cage. This "forced unfolding" mechanism can disrupt misfolded structures, giving the substrate a fresh opportunity to reach its native conformation [33].
Table 1: Comparative Features of Hsp70 and Hsp60 Chaperone Systems
| Feature | Hsp70 System | Hsp60 (GroEL-GroES) System |
|---|---|---|
| Core Components | Hsp70 (DnaK), Hsp40 (DnaJ), NEF (GrpE) | GroEL (Hsp60), GroES (Hsp10) |
| Structural Organization | Monomeric; two-domain structure | Double-ring tetradecamer with a lid |
| ATP Dependency | ATP-dependent allosteric control | ATP-dependent encapsulation |
| Primary Recognition Motif | Short hydrophobic peptide stretches (~7 residues) | Exposed hydrophobic surfaces on non-native proteins |
| Binding Site | Hydrophobic pocket in the SBDβ | Hydrophobic patches on apical domains |
| Folding Environment | Open binding; no encapsulation | Secluded, hydrophilic Anfinsen cage |
| Primary Role in Proteostasis | Holdase; prevents aggregation of nascent/unfolded chains | Foldase; provides isolated compartment for folding |
Experimental Methodologies for Studying Chaperone-Substrate Interactions
Elucidating the dynamic interactions between chaperones and their substrates requires sophisticated biochemical and biophysical techniques. Below are detailed protocols for key experiments cited in research.
Aggregation Prevention Assay
A standard in vitro method to quantify chaperone activity is to monitor its ability to prevent the aggregation of a model substrate under stress conditions [32].
Protocol:
- Substrate Denaturation: A model substrate protein (e.g., citrate synthase or rhodanese) is first denatured in a buffer containing 6 M guanidine-HCl and a reducing agent like DTT [32].
- Refolding Initiation: The denatured substrate is rapidly diluted 100-fold into a refolding buffer. This dilution initiates aggregation as the protein begins to refold and expose hydrophobic surfaces.
- Turbidity Measurement: The sample is placed in a spectrophotometer, and light scattering (turbidity) is monitored by measuring the absorbance at 360 nm for a set period (e.g., 10-30 minutes) at a constant temperature (e.g., 25°C). Aggregation manifests as an increase in optical density.
- Data Analysis: The aggregation kinetics in the presence of the chaperone are compared to a control containing only the substrate. Raw absorbance data are normalized, and relative aggregation is calculated as the fraction of the final absorbance value observed in the control. Effective chaperones will significantly reduce or eliminate the increase in turbidity [32].
READ Method for Visualizing Dynamic Complexes
The Residual Electron and Anomalous Density (READ) method is a novel X-ray crystallography approach designed to visualize heterogeneous and dynamic complexes, such as those between a chaperone and its unfolding substrate [35].
Protocol:
- Complex Crystallization: Co-crystallize the chaperone (e.g., E. coli Spy) with a substrate protein (e.g., Im7).
- Anomalous Scatterer Incorporation: Engineer variants of the substrate where individual residues are replaced with the non-canonical amino acid 4-iodophenylalanine (pI-Phe), a strong anomalous scatterer. Co-crystallize the chaperone with each single-substitute substrate variant [35].
- Data Collection: Collect X-ray diffraction data and anomalous data for each crystal.
- Molecular Dynamics (MD) Simulation: Generate a large, diverse pool of possible chaperone-substrate complex conformations using coarse-grained MD simulations.
- Sample-and-Select Algorithm: A computational algorithm iteratively selects the minimal ensemble of conformations from the MD pool that best fits both the residual electron density and the collected anomalous signals from the iodine atoms.
- Validation: The final structural ensemble is validated using multiple statistical tests, resulting in a high-resolution view of the various conformational states the substrate samples while bound to the chaperone [35].
The following diagram illustrates the logical workflow of the READ method.
Analysis of Hydrophobic Motif Function
To define the role of specific hydrophobic motifs in chaperone activity, such as in BRICHOS or prefoldin domains, researchers employ loop-swap and point mutation strategies [32] [36].
Protocol:
- Sequence Analysis: Identify short, conserved hydrophobic sequence motifs (e.g., tripeptides) in an unstructured loop region of a chaperone domain [36].
- Construct Design:
- Loop-Swap Variants: Replace the native loop in an active chaperone (e.g., Bri2 BRICHOS) with the corresponding loop from a chaperone-inactive homolog (e.g., proSP-C BRICHOS), and vice versa [36].
- Point Mutants: Systematically mutate key hydrophobic residues in the motifs to alanine or more polar residues to reduce hydrophobicity.
- Protein Purification: Express and purify the wild-type and variant chaperone proteins.
- Activity Assay: Test the chaperone activity of all variants using the aggregation prevention assay (see 4.1). The relative activity is quantified and correlated with the biological hydrophobicity of the motifs [36].
- Oligomerization Analysis: Use techniques like analytical size-exclusion chromatography (SEC) to determine if mutations affect the chaperone's assembly state, as oligomerization is often required to display multiple hydrophobic motifs [32] [36].
The Scientist's Toolkit: Key Research Reagents
The following table details essential reagents and materials used in experimental studies of chaperone function.
Table 2: Research Reagent Solutions for Chaperone Studies
| Reagent / Material | Function in Experiment | Key Characteristics & Examples |
|---|---|---|
| Model Amyloidogenic Substrates | To study chaperone inhibition of fibril formation. | Aβ42 (Alzheimer's disease), α-Synuclein (Parkinson's disease). Monitored by Thioflavin T fluorescence [31]. |
| Model Amorphous Aggregation Substrates | To study chaperone prevention of non-fibrillar aggregation. | Citrate Synthase (CS), Rhodanese (Rho), Malate Dehydrogenase (MDH). Unfold at high temperature; aggregation monitored by light scattering [32] [36]. |
| Anomalous Scatterers | To locate specific residues in dynamic complexes via X-ray crystallography. | 4-Iodophenylalanine (pI-Phe): A non-canonical amino acid with a strong anomalous signal for precise positioning [35]. |
| Chaperone Activity Assay Kits | Commercial solutions for standardized activity measurement. | Kits based on light scattering of a standard substrate (e.g., CS or MDH) in the presence of a test chaperone. |
| Size-Exclusion Chromatography (SEC) Columns | To analyze chaperone oligomeric state and complex formation. | Superdex S200HR: Used for analytical SEC to separate oligomers, dimers, and monomers of chaperones like prefoldin and BRICHOS [32] [36]. |
Advanced Research and Therapeutic Implications
Beyond Hydrophobicity: The Role of Electrostatics
While hydrophobic interactions are the primary driving force for substrate recognition, recent research highlights the important role of electrostatic interactions in modulating chaperone activity. For example, engineering the chaperone Spy to increase the positive charge on its surface was shown to enhance its anti-aggregation activity for a variety of substrates. This suggests that attractive electrostatic forces can guide non-native proteins to the chaperone's binding surface, increasing the association rate constant (k~on~) and overall efficiency, without altering the fundamental hydrophobic binding mechanism [37]. This combination of hydrophobic binding and electrostatic steering may be a general feature that enhances the capacity and speed of the cellular chaperone network.
Chaperones in Longevity and Disease
The critical role of chaperones in proteostasis is underscored by their correlation with longevity and involvement in human disease. Comparative studies across vertebrate species have shown that longer-lived mammals and birds consistently exhibit higher constitutive expression levels of Hsp60, Hsp70, GRP78, and GRP94 in tissues like liver, heart, and brain [38]. This suggests that the evolution of longevity is supported by an enhanced proteostasis network with a greater capacity to prevent protein damage and aggregation over a longer lifespan [38]. Conversely, a decline in chaperone function is implicated in neurodegenerative diseases such as Alzheimer's and Parkinson's disease. Here, chaperones like Hsp70 and small HSPs can interact with amyloidogenic proteins (e.g., Aβ, α-synuclein) at various stages of the aggregation pathway, inhibiting primary nucleation, secondary nucleation, and elongation, or even mediating the detoxification of amyloid fibrils [31].
Targeting Chaperones for Drug Discovery
The clear link between chaperone malfunction and disease has positioned them as promising therapeutic targets. The field has evolved through several stages:
- Stage 1 (Pan-isoform inhibition): Development of small-molecule inhibitors (e.g., for Hsp90) without initial regard for isoform selectivity.
- Stage 2 (Isoform-selective inhibition): Designing inhibitors that target specific isoforms of HSP families (e.g., cytosolic HSP90α vs. ER-resident GRP94) to improve specificity and reduce side effects.
- Stage 3 (Targeting PPIs): Developing compounds that disrupt specific protein-protein interactions (PPIs) between HSPs and their co-chaperones or client proteins.
- Stage 4 (Multi-specific molecules): The latest frontier involves designing bi- or multi-specific molecules that simultaneously target different components of the chaperone network for enhanced efficacy and selectivity [6].
The following diagram illustrates the progression of these therapeutic strategies.
Cellular compartmentalization, a defining feature of eukaryotic cells, establishes distinct biochemical environments within membrane-bound organelles, allowing separate metabolic reactions and specialized functions to occur simultaneously and efficiently. This physical separation minimizes conflicting side reactions, prevents accidental enzyme inhibition, and enables cells to increase internal surface areas for critical reactions through organelle membranes [39]. The functional specialization achieved through compartmentalization is fundamentally supported by molecular chaperones, which ensure proper protein folding, assembly, and quality control within each cellular compartment [16].
The proteostasis network—an integrated system of molecular chaperones, folding enzymes, and degradation machineries—maintains protein homeostasis differently in each compartment [16]. Molecular chaperones, particularly those from the heat shock protein (HSP) families, have evolved specialized forms and regulatory mechanisms tailored to the unique environments and client proteins of the cytosol, mitochondria, and endoplasmic reticulum. This review examines the compartment-specific functions of the Hsp70 and Hsp60 chaperone systems, their mechanisms of action, and their critical roles in maintaining cellular proteostasis.
Compartment-Specific Chaperone Systems
The Cytosolic Hsp70 Chaperone Network
The 70-kDa heat shock protein (Hsp70) chaperone system in the cytosol represents a paradigm of "selective promiscuity," interacting with a wide range of substrate proteins while minimizing undesired interactions [40]. Cytosolic Hsp70 facilitates the folding, assembly, membrane translocation, and quality control of nascent polypeptide chains and stress-denatured proteins through an ATP-dependent binding and release cycle.
Mechanism and Regulation: Hsp70 achieves its functional versatility through sophisticated regulation by co-chaperones. J-domain proteins (JDPs) and nucleotide exchange factors (NEFs) are key to substrate recognition, remodeling, and release from chaperone complexes [40]. The approximately 50 human JDPs confer remarkable client specificity to Hsp70s, with some JDPs targeting Hsp70s to specific subcellular locations ("recruiters") while others bind substrates directly as "specialists" or "generalists" [40]. Different JDPs, together with NEFs, dictate the fate of Hsp70 clients by directing them toward distinct protein quality control pathways, resulting in either folding or degradation [40].
Functional Specialization: The cytosolic Hsp70 system demonstrates remarkable adaptation to its environment through:
- Collaboration with other chaperones: Hsp70 works with Hsp90 in multi-chaperone complexes for folding specific client classes, such as kinases and transcription factors [6].
- Integration with degradation pathways: Through co-chaperones like BAG-1, Hsp70 can target irreversibly damaged proteins for proteasomal degradation [41].
- Stress adaptation: Under heat stress, Hsp70 expression increases dramatically to prevent aggregation of denatured proteins and facilitate refolding [41].
Table 1: Key Components of the Cytosolic Hsp70 System
| Component | Type | Function | Regulatory Role |
|---|---|---|---|
| Hsp70 (HSPA1A, HSPA8) | Core Chaperone | ATP-dependent substrate binding/release | Protein folding, translocation, complex assembly |
| Hsp40 (DNAJA, DNAJB, DNAJC) | J-domain Protein | Stimulates Hsp70 ATPase activity | Substrate targeting and specificity |
| BAG-1, BAG-3 | Nucleotide Exchange Factor | Promotes ADP release from Hsp70 | Determines client fate (degradation vs. folding) |
| Hsp110 | NEF | Specialized NEF for Hsp70 | Enhances folding efficiency under stress |
| CHIP | Co-chaperone | E3 ubiquitin ligase | Targets Hsp70 clients for degradation |
Mitochondrial Chaperone Systems: Hsp60 and Hsp70
Mitochondria contain specialized chaperone systems in different sub-compartments that are essential for maintaining mitochondrial proteostasis and overall function. The mitochondrial matrix houses the Hsp60 chaperonin system, while both the matrix and intermembrane space contain specialized Hsp70 isoforms.
Mitochondrial Hsp60 (HSPD1) Structure and Function: The Hsp60 chaperonin forms a large double-ring complex with a central folding cavity that provides an isolated environment for protein folding. Unlike Hsp70, which primarily binds extended hydrophobic segments, Hsp60 encapsulates entire folding intermediates, preventing aggregation during the folding process [42]. Hsp60 deficiency disrupts the mitochondrial matrix proteome and has been shown to dysregulates cholesterol synthesis, demonstrating its essential role in mitochondrial function and metabolism [42].
Mitochondrial Hsp70 System: Mitochondrial Hsp70 (mtHsp70) plays distinct roles in the matrix, including:
- Protein import: mtHsp70 is essential for the import of nuclear-encoded proteins through the TIM complex, using ATP-dependent binding to pull precursors into the matrix.
- Folding of matrix proteins: Once imported, mtHsp70 assists in the folding of proteins in collaboration with the Hsp60/Hsp10 system.
- Proteostasis maintenance: mtHsp70 works with mitochondrial proteases to ensure degradation of misfolded proteins.
Integration with Mitochondrial Quality Control: Mitochondrial chaperones are integral components of the mitochondrial quality control (MQC) system, which includes mitochondrial dynamics (fusion and fission), mitophagy, and proteostasis [43]. This system maintains a healthy mitochondrial population through:
- Protein quality control: Molecular chaperones like HSP70 and HSP90 assist in protein folding, while degradation systems including Lon and ClPXP proteases remove misfolded or damaged proteins [44].
- Coordination with dynamics: Mitochondrial fusion allows content mixing, complementing chaperone function by enabling functional complementation between damaged mitochondria [43].
- Mitophagy regulation: When chaperones cannot rescue damaged proteins, the MQC system triggers mitophagy to remove severely damaged mitochondria [43].
Table 2: Mitochondrial Chaperone Systems and Their Functions
| Chaperone | Location | Complex Structure | Primary Functions | Associated Diseases |
|---|---|---|---|---|
| Hsp60 (HSPD1) | Matrix | Tetradecameric double-ring | Folding of matrix proteins, metabolic regulation | Neurodegeneration, myelination defects |
| mtHsp70 (HSPA9) | Matrix | Monomeric with co-chaperones | Protein import, folding, iron-sulfur cluster biogenesis | Myelodysplastic syndromes |
| TRAP-1 | Matrix | Homodimer | Mitochondrial unfolded protein response, protection from oxidative stress | Cancer, neurodegenerative diseases |
| HSP10 | Matrix | Single heptameric ring | Co-chaperone for Hsp60, facilitates substrate release | Neurodevelopmental disorders |
Endoplasmic Reticulum Chaperone Systems
The endoplasmic reticulum (ER) contains specialized chaperone systems adapted for the folding of secretory and membrane proteins, which face unique challenges including disulfide bond formation, glycosylation, and coping with fluctuating calcium levels.
ER Hsp70 (BiP) Structure and Function: The ER-resident Hsp70 family member, Binding immunoglobulin Protein (BiP), serves as the central chaperone in the ER lumen. BiP performs multiple essential functions:
- Recognition of hydrophobic patches: BiP binds exposed hydrophobic regions on unfolded proteins, preventing aggregation.
- Translocation: BiP seals the translocon pore when not actively importing proteins, preventing calcium leakage.
- ER-associated degradation (ERAD): BiP identifies persistently misfolded proteins for retrotranslocation and degradation.
- UPR activation: BiP acts as the master regulator of the unfolded protein response (UPR); accumulation of unfolded proteins causes BiP release from UPR sensors, activating signaling pathways.
Collaboration with ER-Specific Co-chaperones: ER Hsp70 functions with specialized J-domain proteins that are integrated into various ER processes. These include:
- ERdj3: A soluble JDP that delivers clients to BiP.
- ERdj4: A membrane-associated JDP involved in ERAD substrate recognition.
- ERdj5: An oxidoreductase-containing JDP that reduces disulfide bonds on misfolded proteins prior to their retrotranslocation.
Integration with ER Quality Control: The ER chaperone network operates within a comprehensive quality control system that includes:
- Calnexin/Calreticulin cycle: For glycoprotein folding.
- Protein disulfide isomerases: For correct disulfide bond formation.
- ER degradation-enhancing α-mannosidase-like proteins: For recognition of terminally misfolded glycoproteins.
Table 3: Endoplasmic Reticulum Chaperone Systems
| Chaperone | Type | Location in ER | Key Functions | Regulatory Mechanisms |
|---|---|---|---|---|
| BiP (HSPA5) | Hsp70 | Lumen | General protein folding, translocation seal, UPR regulation | ATP/ADP cycle, J-domain proteins |
| GRP94 | Hsp90 | Lumen | Folding of secreted proteins, integrins, toll-like receptors | ATP-dependent, Ca²⁺ binding |
| Calnexin | Lectin Chaperone | Membrane | Glycoprotein folding, quality control | Recognition of monoglucosylated N-glycans |
| Calreticulin | Lectin Chaperone | Lumen | Glycoprotein folding, Ca²⁺ storage | Similar to calnexin but soluble |
| ERdj3 | J-domain Protein | Lumen | BiP co-chaperone, client delivery | Substrate recognition and targeting |
Experimental Analysis of Chaperone Functions
Methodologies for Studying Compartment-Specific Chaperone Mechanisms
Proteomic Analysis of Chaperone Deficiencies: Recent advances in proteomic technologies enable comprehensive analysis of chaperone functions. As demonstrated in Hsp60 deficiency studies, researchers employ multi-omics approaches including:
- Quantitative mass spectrometry: To measure changes in the mitochondrial matrix proteome following chaperone depletion [42].
- Transcriptomic profiling: To identify compensatory pathways activated in response to chaperone deficiency.
- Metabolomic analysis: To detect metabolic consequences of disrupted mitochondrial proteostasis, such as dysregulated cholesterol synthesis observed in Hsp60 deficiency [42].
Protocol: Proteomic Analysis of Hsp60-Deficient Mitochondria
- Genetic manipulation: Create Hsp60-deficient models using CRISPR/Cas9 in zebrafish or mammalian cell lines.
- Mitochondrial isolation: Purify mitochondria using differential centrifugation and density gradient purification.
- Protein extraction and digestion: Lyse mitochondria, extract proteins, and digest with trypsin.
- LC-MS/MS analysis: Perform liquid chromatography-tandem mass spectrometry with isobaric labeling for quantification.
- Bioinformatic analysis: Identify significantly altered proteins and pathways using specialized software.
- Validation: Confirm key findings using Western blotting or targeted assays.
Functional Assays for Chaperone Activity: Researchers employ various biochemical and cell-based assays to assess chaperone function:
- ATPase activity assays: Measure the ATP hydrolysis rate of Hsp70 chaperones.
- Substrate refolding assays: Monitor the recovery of enzymatic activity from denatured substrates.
- Aggregation suppression assays: Measure the ability of chaperones to prevent aggregation of thermolabile substrates.
- Protein import assays: Specifically for mitochondrial chaperones, assess import efficiency of precursor proteins.
Visualization of Chaperone Networks and Pathways
Diagram 1: Hsp70 Chaperone Cycle Across Cellular Compartments. The core ATPase cycle of Hsp70 chaperones is conserved but utilizes compartment-specific co-chaperones: cytosolic Hsp70 works with Hsp40 and BAG proteins; mitochondrial mtHsp70 (HSPA9) collaborates with the TIM complex and Mge1; ER-resident BiP (HSPA5) functions with ERdj proteins and GRP170.
Diagram 2: Mitochondrial Quality Control Network. Mitochondrial chaperones (HSP60, mtHSP70) function within an integrated quality control system that includes dynamics (fusion/fission), proteostasis (chaperones and proteases), biogenesis (PGC-1α pathway), and clearance mechanisms (mitophagy).
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for Chaperone Studies
| Reagent/Category | Specific Examples | Function/Application | Compartment Specificity |
|---|---|---|---|
| Inhibitors | PES (phenylethynesulfonamide) | HSP70 inhibitor; blocks T817MA-mediated neuroprotection [45] | Cytosolic/Mitochondrial |
| Mdivi-1 | DRP1 inhibitor; blocks mitochondrial fission [43] | Mitochondrial | |
| 17-AAG | HSP90 inhibitor; used in cancer studies [6] | Multiple compartments | |
| Animal Models | Zebrafish HSP60 mutants | Model for mitochondrial chaperonopathy [42] | Mitochondrial |
| MCAO mouse model | Middle cerebral artery occlusion; studies brain ischemia [45] | Multiple compartments | |
| Cell Models | HT22 cells (hippocampal) | Oxygen-glucose deprivation studies [45] | Multiple compartments |
| OGD model | Oxygen-glucose deprivation; mimics ischemia [45] | Multiple compartments | |
| Antibodies | Anti-HSP70 | Detects induced HSP70 in Western blot, IHC [45] | Multiple compartments |
| Anti-HSP60 | Mitochondrial matrix chaperone detection [42] | Mitochondrial | |
| Anti-CHOP | ER stress marker detection [45] | Endoplasmic Reticulum | |
| Expression Systems | Recombinant HSP70/HSP60 | Protein-protein interaction studies, ATPase assays | Compartment-specific isoforms |
| Chemical Inducers | T817MA compound | Upregulates HSP70, downregulates HSP90 [45] | Multiple compartments |
Implications for Therapeutic Development
The compartment-specific functions of molecular chaperones present unique opportunities for therapeutic intervention in various diseases. The neuroprotective effects of T817MA in brain ischemia models demonstrate the therapeutic potential of modulating chaperone networks. This compound attenuates oxidative stress and ER stress via the HSP70-HSP90 pathway, significantly improving cell viability and reducing reactive oxygen species production in OGD-treated HT22 cells and MCAO mouse models [45]. Critically, the protection afforded by T817MA is abolished by the HSP70 inhibitor PES, confirming the essential role of the HSP70-HSP90 pathway in its mechanism of action [45].
Strategic Considerations for Chaperone-Targeted Therapies:
- Isoform selectivity: Development of inhibitors targeting specific HSP90 isoforms (cytosolic HSP90α/β, ER-resident GRP94, mitochondrial TRAP-1) to minimize off-target effects [6].
- Protein-protein interaction targeting: Disruption of specific chaperone-co-chaperone interactions represents a more precise approach than pan-inhibition [6].
- Combinatorial approaches: Leveraging chaperone inhibitors to sensitive cells to other therapeutics, particularly in cancer and neurodegenerative diseases.
- Adaptation to cellular stress: Utilizing the elevated chaperone requirements of cancer cells for selective targeting while sparing normal cells.
The intricate compartmentalization of chaperone systems underscores the importance of understanding their distinct mechanisms and interactions for developing effective therapeutic strategies that restore proteostasis in disease states.
Cellular compartmentalization enables specialized chaperone systems in the cytosol, mitochondria, and endoplasmic reticulum to maintain proteostasis through tailored mechanisms. The Hsp70 system demonstrates remarkable adaptability across compartments, while specialized chaperones like Hsp60 fulfill unique roles in organelles with distinct folding environments. These compartment-specific networks operate within integrated quality control systems that balance protein folding, degradation, and organelle dynamics. Continued elucidation of these mechanisms, aided by advanced proteomic and structural approaches, promises new therapeutic strategies for diseases of proteostasis disruption, from neurodegeneration to cancer. The sophisticated regulation of chaperone networks across cellular compartments highlights both the challenges and opportunities for targeted intervention in proteostasis-related diseases.
Advanced Research Methodologies and Therapeutic Targeting Strategies
Protein homeostasis, or proteostasis, is a fundamental cellular process that ensures proteins are correctly synthesized, folded, trafficked, and degraded. The Hsp70 and Hsp60 chaperone families are central to this network, preventing toxic aggregation and facilitating the recovery of misfolded proteins—processes critically implicated in neurodegenerative diseases, cancer, and aging [46] [15]. Understanding the mechanistic actions of these chaperones requires atomic-level insights into their complex conformational dynamics and client interactions. Structural biology provides this window, with X-ray crystallography and cryo-electron microscopy (cryo-EM) emerging as the two dominant, complementary techniques for elucidating chaperone mechanisms [47]. This technical guide examines the principles, methodologies, and applications of these techniques, framed within the context of contemporary proteostasis research on Hsp70 and Hsp60 systems, providing a resource for scientists engaged in targeted therapeutic development.
Technical Foundations of High-Resolution Structure Determination
X-ray Crystallography: Principles and Workflow
X-ray crystallography determines atomic structures by measuring how X-rays diffract from highly ordered crystalline lattices of biomolecules. The foundational principle is Bragg's Law, which describes the relationship between the diffraction angles and the spacing of crystal planes [47].
Key Steps in the Experimental Pipeline:
- Protein Purification and Crystallization: The target chaperone (e.g., Hsp70 or a subcomplex) is purified to homogeneity. Crystallization involves screening vast numbers of conditions to find the precise chemical parameters that promote the formation of well-ordered, three-dimensional crystals. This often requires significant quantities of protein and can be the rate-limiting step [47].
- Data Collection: A crystal is exposed to an intense X-ray beam, producing a diffraction pattern of discrete spots. The quality of this pattern determines the achievable resolution.
- Phase Determination: The "phase problem" is solved using methods like molecular replacement (using a known homologous structure), or experimental techniques such as Single-wavelength Anomalous Dispersion (SAD/MAD) [47].
- Model Building and Refinement: An initial atomic model is built into the experimental electron density map and iteratively refined against the diffraction data to achieve the best possible fit [47].
Cryo-Electron Microscopy: Principles and Workflow
Cryo-EM visualizes macromolecules frozen in a thin layer of vitreous ice, preserving their native state. Single-particle analysis (SPA) involves computationally aligning and averaging images of thousands of individual particles to reconstruct a high-resolution 3D structure [47].
Key Steps in the Experimental Pipeline:
- Sample Vitrification: The purified chaperone complex solution is applied to a grid and rapidly plunged into a cryogen (typically liquid ethane), freezing it so quickly that water molecules do not have time to crystallize. This embeds the particles in a glass-like ice layer [47].
- Data Acquisition: The vitrified grid is imaged in a transmission electron microscope under low-dose conditions to minimize radiation damage. Thousands to millions of micrographs are collected, each containing images of the complex in random orientations [48].
- Image Processing: This critical step involves particle picking, 2D classification to group similar particle views, and 3D reconstruction to generate an initial density map. Often, multiple rounds of 3D classification are used to separate structural heterogeneities (e.g., different conformational states of Hsp70) [47].
- Model Building and Refinement: An atomic model is built de novo or from a homologous structure and refined against the cryo-EM density map [47].
Table 1: Quantitative Comparison of X-ray Crystallography and Cryo-EM for Chaperone Studies
| Feature | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Typical Resolution | Atomic (often ≤1.5 Å) to medium (~3 Å) | Near-atomic (≤3 Å) to low resolution (>10 Å) [47] |
| Sample Requirement | High purity and concentration; large quantity for screening | High purity; lower total quantity required [47] |
| Key Sample Challenge | Growing well-diffracting 3D crystals [48] | Sample heterogeneity and preferred orientation on grids |
| Sample State | Packed, crystalline lattice | Solution state, near-native environment [47] |
| Data Analysis | Crystallographic software (e.g., PHENIX) | Complex computational processing (e.g., RELION, cryoSPARC) |
| Throughput | Medium to high (after crystal optimization) | Increasingly high with automation |
| Ideal for | High-resolution snapshots of stable complexes and domains | Large, flexible complexes, multiple conformations, membrane proteins [48] |
Experimental Protocols for Chaperone Complex Analysis
Protocol 1: Determining an Hsp70-Client Complex Structure via Cryo-EM
This protocol is adapted from recent studies that solved the full-length structures of the Hsp70-Hsp40-client complex [49] [50].
1. Complex Reconstitution: - Purify full-length Hsp70 (e.g., DnaK), its Hsp40 co-chaperone (e.g., DnaJ), and a model client peptide. - Incubate the components in a stoichiometric ratio (e.g., 2:2:1 Hsp70:Hsp40:client) in the presence of a non-hydrolyzable ATP analog (e.g., ATPγS) to trap the active state. Include Mg²⁺ and K⁺ ions as they are essential for Hsp70's ATPase activity [51].
2. Sample Vitrification: - Apply 3-4 µL of the complex (at ~0.5-1 mg/mL) to a freshly glow-discharged cryo-EM grid. - Blot away excess liquid and plunge-freeze the grid in liquid ethane using a vitrification device (e.g., Vitrobot). Optimize blotting time and humidity to achieve a thin, homogeneous ice layer.
3. Data Collection: - Load the grid into a 300 kV cryo-electron microscope equipped with a direct electron detector (e.g., K3 or Falcon 4) and a energy filter. - Collect a large dataset (e.g., 5,000-10,000 micrographs) automatically using software like SerialEM or EPU. Use a defocus range of -0.5 to -2.5 µm and a total electron dose of ~40-60 e⁻/Ų.
4. Image Processing and 3D Reconstruction: - Pre-process micrographs: patch motion correction, estimate and correct for CTF (Gctf, CTFFIND-4). - Use template-based or AI-driven picking (e.g., in cryoSPARC or RELION) to extract ~1-5 million particles. - Perform several rounds of 2D classification to remove junk particles. - Generate an initial model ab initio or via homology modeling, then use heterogeneous refinement (3D classification) to separate distinct conformational states of the chaperone complex. - Refine the selected classes to high resolution using non-uniform refinement and perform post-processing to sharpen the map and correct for modulation transfer function (MTF) and B-factor.
5. Atomic Model Building: - If a high-resolution map (better than ~3.5 Å) is obtained, build an atomic model de novo using Coot. - For lower-resolution maps, dock existing crystal structures of Hsp70 domains (e.g., PDB: 1DKZ) and Hsp40 (e.g., PDB: 1XBL) into the density as rigid bodies using UCSF Chimera or Situs [47]. - Refine the model against the cryo-EM map using real-space refinement in PHENIX or ISOLDE, with geometry and stereochemical restraints.
Protocol 2: Hybrid Approach for Large Chaperone Assemblies
For large, heterogeneous complexes like the Hsp60 chaperonin (GroEL-GroES), a hybrid approach is often necessary.
1. Cryo-EM for the Holocomplex: - Follow a protocol similar to 3.1 to obtain a medium-resolution (4-8 Å) cryo-EM map of the entire GroEL-GroES complex.
2. X-ray Crystallography of Subcomplexes: - Express, purify, and crystallize individual domains or subcomplexes of GroEL and GroES. - Solve their atomic structures using X-ray crystallography, potentially with molecular replacement using existing homologous structures.
3. Integrative Modeling: - Rigid-body dock the high-resolution X-ray structures of the subcomplexes into the lower-resolution cryo-EM envelope of the full complex [47]. - Use flexible fitting algorithms, such as molecular dynamics flexible fitting (MDFF) or normal mode analysis (NMA), to adjust the atomic models to better fit the cryo-EM density, accounting for conformational changes upon assembly [47].
Research Reagent Solutions for Structural Studies of Chaperones
Table 2: Essential Reagents and Materials for Structural Studies of Hsp70/Hsp60 Complexes
| Reagent / Material | Function in Experiment | Example & Notes |
|---|---|---|
| Recombinant Chaperones | The primary target for structural analysis. | Human Hsp70 (HSPA1A), Hsp40 (DNAJA1), Hsp60. Requires high-purity (>95%) for crystallization/cryo-EM. |
| Co-chaperones & Clients | To form functional complexes for study. | Nucleotide Exchange Factors (NEFs like Bag-1), model client peptides (e.g., NRLLLTG) [51]. |
| ATP/ATPγS/ADP | To study chaperone function in specific nucleotide states. | ATPγS (a non-hydrolyzable analog) traps the ATP-bound state; ADP stabilizes the high-affinity state. |
| Lipids & Detergents | For membrane-associated chaperones or crystallography. | DDM, LMNG for solubilizing membrane protein clients; lipids for lipidic cubic phase (LCP) crystallization. |
| Crystallization Screens | To identify initial conditions for crystal formation. | Commercial sparse matrix screens (e.g., from Hampton Research, Molecular Dimensions). |
| Cryo-EM Grids | The support substrate for vitrified samples. | UltrAuFoil (gold) or Quantifoil (copper) grids with various hole sizes and carbon support films. |
| Negative Stain | For rapid sample quality assessment and grid screening. | Uranyl acetate or phosphotungstic acid. Provides quick, low-resolution feedback. |
| Chaperone-Specific Antibodies | For affinity purification and functional assays. | Anti-Hsp70, anti-Hsp60; used for pull-downs or Western blotting to verify complex formation. |
Data Integration and Visualization in Chaperone Research
Structural biology is increasingly integrative. X-ray crystallography and cryo-EM are not mutually exclusive but are powerfully combined. A common workflow uses a low-resolution cryo-EM map of a large chaperone complex as a scaffold to dock high-resolution X-ray structures of its components, a practice known as rigid-body docking [47]. For example, the crystal structure of the Ryanodine receptor's SPRY2 domain was docked into a 10 Å cryo-EM map with high precision, a result later confirmed by a high-resolution cryo-EM structure [47]. Furthermore, flexible docking algorithms like MDFF and Flex-EM allow for conformational adjustments of the X-ray models to fit the cryo-EM density, revealing interaction interfaces and conformational changes [47].
The following diagram illustrates the complementary workflow for determining the structure of a chaperone complex using an integrative approach:
Integrative Structural Biology Workflow
The synergistic application of X-ray crystallography and cryo-EM is pivotal for advancing our understanding of chaperone-mediated proteostasis. While crystallography continues to provide unmatched resolution for stable structures, cryo-EM has revolutionized the study of large, dynamic chaperone complexes in near-native states [47] [48]. The recent elucidation of the full-length Hsp70-Hsp40 complex structure, achieved by integrating cryo-EM with other biophysical techniques, underscores the power of this hybrid approach [49] [50]. It revealed the autoinhibitory mechanism of Hsp40 and the precise role of its G/F-rich region as a pseudo-substrate for client handoff to Hsp70—a key mechanistic insight with direct implications for diseases of proteostasis failure [49] [50]. For researchers in drug discovery, these structural insights create a foundation for developing small-molecule modulators of chaperone function. As both techniques continue to evolve, particularly with advancements in time-resolved methods [52], our capacity to visualize the intricate dance of chaperones and their clients will undoubtedly lead to novel therapeutic strategies for cancer, neurodegeneration, and other protein misfolding diseases.
Biochemical Assays for Monitoring ATPase Cycles and Client Interactions
In the intricate machinery of cellular proteostasis, molecular chaperones such as Hsp70 and Hsp60 are indispensable components that prevent protein misfolding and aggregation. The ability of these chaperones to fold nascent polypeptides, refold denatured proteins, and direct damaged proteins for degradation is fundamentally powered by adenosine triphosphate (ATP) hydrolysis [53]. ATPase activity—the enzymatic conversion of ATP to adenosine diphosphate (ADP) and inorganic phosphate (Pi)—drives the conformational changes in chaperones necessary for their client-protein interactions [19] [53]. Consequently, precise monitoring of ATPase cycles is not merely a biochemical exercise but a crucial window into understanding chaperone function, regulation, and mechanism.
This technical guide details the core assays for quantifying ATPase activity and analyzing client interactions, with a specific focus on the Hsp70 and Hsp60 chaperone families. These assays form the bedrock of research aimed at deciphering how chaperones maintain protein homeostasis, and they are increasingly vital in drug discovery campaigns targeting chaperones implicated in cancer and neurodegenerative diseases [19] [54]. We present both foundational and advanced methodologies, structured data for easy comparison, and essential resources to equip researchers with the tools for rigorous mechanistic investigation.
Chaperone ATPase Mechanisms: Hsp70 and Hsp60
Hsp70 ATPase Cycle
The Hsp70 chaperone system is a paradigm of allosteric regulation. Hsp70 consists of a nucleotide-binding domain (NBD) and a substrate-binding domain (SBD) [19]. The ATPase cycle is critically regulated by co-chaperones: J-domain proteins (JDP/Hsp40) stimulate the inherently slow ATP hydrolysis, while nucleotide exchange factors (NEFs) facilitate the replacement of ADP with ATP [19].
- ATP-bound State: In this state, Hsp70 exhibits a low affinity for client proteins, resulting in fast binding and release kinetics. This "open" conformation allows clients to bind and be rapidly released [19].
- ADP-bound State: ATP hydrolysis triggers a conformational shift to a high-affinity "closed" state, characterized by slow client binding and release. This stabilizes the interaction with the client protein, providing a protected environment for folding [19].
The cycle continues as NEFs promote ADP release, allowing ATP to rebind and the client to be released in a folded, or more folded, state [19].
Hsp60 (Chaperonin) ATPase Cycle
The Hsp60 family, or chaperonins, form large double-ring complexes (e.g., GroEL in bacteria, TRiC in eukaryotes) that provide an isolated chamber for protein folding [53].
- Client Binding: Non-native client proteins bind to hydrophobic patches in the apical domains of the chaperonin ring [53].
- ATP-Driven Encapsulation: ATP binding and hydrolysis trigger large conformational changes. In GroEL, this leads to binding of the co-chaperone GroES, encapsulating the client within the folding chamber. TRiC undergoes similar conformational changes without a separate co-chaperone [53].
- Folding and Release: The client protein folds in isolation for the duration of the ATP hydrolysis cycle, which helps prevent aggregation. After hydrolysis, the client is released; if folding is incomplete, the client may rebind for another attempt [53].
Table 1: Key Characteristics of Hsp70 and Hsp60 Chaperone Systems.
| Feature | Hsp70 | Hsp60 (Chaperonins) |
|---|---|---|
| Structure | Monomeric with NBD and SBD | Large double-ring complex (e.g., GroEL: 7 subunits/ring; TRiC: 8 subunits/ring) |
| Primary Folding Mechanism | Transient binding and release, preventing aggregation | ATP-driven encapsulation in an isolated chamber |
| Role of ATP Hydrolysis | Drives conformational change between high/low client affinity states | Powers large-scale conformational changes for client encapsulation and release |
| Key Co-chaperones | J-domain proteins (Hsp40), Nucleotide Exchange Factors (NEFs) | GroES (for Group I) |
| Client Specificity | Broad, recognizes hydrophobic peptides | Broad, but TRiC has subunit heterogeneity for defined client orientation |
The following diagram illustrates the core ATPase-driven functional cycles of Hsp70 and Hsp60 chaperonins.
Core ATPase Activity Assays
Quantifying the rate of ATP hydrolysis is fundamental to characterizing chaperone function. The following assays measure the inorganic phosphate (Pi) or ADP produced by the enzymatic reaction.
Malachite Green Phosphate Assay
This classic colorimetric endpoint assay is based on the formation of a green complex between malachite green molybdate and inorganic phosphate, measurable at 650 nm [55].
Detailed Protocol [55]:
- Reaction Setup:
- Combine purified ATPase (e.g., 0.25–5 µM final concentration) with ATP (e.g., 100 mM stock in Tris base) in an appropriate assay buffer (e.g., 100 mM HEPES pH 8.5, 65 mM NaCl, 5% glycerol).
- Include MgCl₂ or another required metal cofactor (e.g., 100 mM stock, premixed 1:1 with ATP).
- Always include a negative control with no enzyme to account for non-enzymatic ATP hydrolysis.
Incubation and Sampling:
- Incubate the reaction at the desired temperature (e.g., 37°C).
- Remove aliquots (e.g., 5 µL) at regular intervals (e.g., 0, 15, 30, 45, 60 min) and immediately dilute them 1:50 in assay buffer to stop the reaction.
- Flash-freeze diluted samples in a dry ice/ethanol bath and store at -80°C until analysis.
Phosphate Detection:
- Thaw samples and add them to a 96-well plate.
- Prepare a phosphate standard curve by serial dilution (e.g., from 40 µM to 0 µM).
- Add malachite green detection reagent (100 µL) to each well, mix, and incubate for 25 minutes at room temperature.
- Measure the absorbance at 650 nm using a microplate reader.
Data Analysis:
- Calculate phosphate concentration for each sample using the standard curve equation.
- Subtract the time-zero value to determine enzyme-specific Pi release.
- Plot nmol Pi/µmol protein versus time. The slope of the linear fit represents the ATPase activity (nmol Pi/µmol protein/min) [55].
Fluorescence-Based ADP Detection Assays
Modern high-throughput screening (HTS) often employs homogeneous, fluorescence-based assays that directly detect ADP, offering superior sensitivity and safety.
Transcreener ADP² Assay Principle [54]: This platform uses a competitive immunoassay format. A fluorescently labeled ADP tracer binds to an anti-ADP antibody, resulting in a high polarization (FP) or TR-FRET signal. As the ATPase reaction produces ADP, it displaces the tracer from the antibody, causing a measurable decrease in FP or a shift in the TR-FRET ratio.
Typical Procedure [54]:
- Reaction Setup: Combine purified ATPase with ATP substrate in a suitable buffer.
- Incubation: Allow hydrolysis to proceed for a defined time.
- Detection: Add a detection mix containing the antibody and fluorescent tracer.
- Signal Measurement: Read fluorescence using an FP, FI, or TR-FRET compatible plate reader.
- Data Analysis: Calculate ADP concentration from the standard curve to determine enzyme velocity and inhibitor potency.
Table 2: Comparison of Core ATPase Activity Assay Methods.
| Assay Parameter | Malachite Green Phosphate Assay | Fluorescence-Based ADP Detection |
|---|---|---|
| Detection Target | Inorganic Phosphate (Pi) | ADP |
| Readout Mode | Colorimetric (Absorbance) | Fluorescence Polarization (FP), TR-FRET, or Fluorescence Intensity (FI) |
| Throughput | Low to medium (endpoint or kinetic) | High (HTS compatible) |
| Key Advantage | Cost-effective; direct measurement of a hydrolysis product. | Homogeneous ("mix-and-read"); no coupling enzymes; highly sensitive; adaptable to multiple formats. |
| Key Disadvantage | Susceptible to interference from phosphate contaminants or compounds [55]. | Higher reagent cost. |
| Ideal Application | Initial enzyme characterization; low-budget studies. | High-throughput inhibitor screening; detailed kinetic profiling. |
Advanced Applications: Monitoring Client Interactions
Beyond basal ATPase rates, a key functional metric is the modulation of this activity by client proteins and co-chaperones. For instance, the bacterial AAA+ protease ClpXP exhibits changes in ATP hydrolysis upon binding to its peptidase partner ClpP, a phenomenon also observable in chaperone systems [56].
Experimental Approach:
- Titrate the client protein or co-chaperone (e.g., Hsp40 for Hsp70) into the ATPase reaction mixture.
- Monitor the resulting change in ATPase rate. This can manifest as activation (e.g., J-domain proteins stimulating Hsp70's ATP hydrolysis [19]) or repression (e.g., ClpP repressing ClpX ATPase activity [56]).
- Apparent binding affinities (Kd) can be determined by plotting the ATPase rate against the client/co-chaperone concentration and fitting the data to a binding model [56].
Table 3: Quantitative Data from ATPase-Client Interaction Studies (Exemplar).
| Chaperone/ATPase | Interacting Partner | Effect on ATPase Rate | Apparent Affinity (Kd) | Assay Method |
|---|---|---|---|---|
| ClpX-ΔN (WWW/WWW) | ClpP tetradecamer | -49% repression | 50 - 170 nM [56] | ATPase inhibition, Protease activity, Peptidase activity |
| Hsp70 | Hsp40 (JDP) | Activation (Increased rate) | Varies by specific partner [19] | Coupled enzyme or ADP detection |
The following diagram outlines a generalized workflow for conducting and analyzing these interaction studies.
Successful execution of these assays requires high-quality reagents and optimized tools. The following table catalogues key materials.
Table 4: Research Reagent Solutions for ATPase and Client Interaction Studies.
| Reagent / Material | Function / Description | Example / Key Considerations |
|---|---|---|
| Purified Chaperone/ATPase | The enzyme of interest. | Hsp70, Hsp60, ClpX; Requires high purity and proper folding; often needs oligomerization for full activity [56] [55]. |
| ATP Substrate | Primary substrate for the reaction. | High-purity ATP; prepare fresh stocks in Tris base to avoid acid-catalyzed hydrolysis; aliquot and store at -20°C [55]. |
| MgCl₂ (or other metals) | Essential cofactor for most ATPases. | Typically added as Mg²⁺; concentration must be optimized as it can affect kinetics [55]. |
| Malachite Green Reagent | Colorimetric detection of inorganic phosphate. | Commercial kits available; sensitive to phosphate contamination in water and labware [55]. |
| Transcreener ADP² Assay Kit | Fluorescence-based detection of ADP. | BellBrook Labs; Available in FP, FI, and TR-FRET formats for HTS; direct detection avoids coupled enzymes [54]. |
| 96-/384-Well Plates | Reaction vessel for assays. | Use plates compatible with the detection method (e.g., clear bottom for absorbance, black for fluorescence). |
| Microplate Reader | Instrument for signal detection. | Absorbance reader (650 nm) for malachite green; fluorescence reader (with FP or TR-FRET capabilities) for ADP detection. |
Biochemical assays for ATPase activity and client interactions are powerful, indispensable tools for dissecting the molecular mechanisms of Hsp70, Hsp60, and other ATP-dependent chaperones. From the traditional malachite green assay to modern HTS-compatible fluorescence platforms, these methods provide quantitative insights into enzymatic kinetics, allosteric regulation, and protein-protein interactions. As research continues to link chaperone dysfunction to human disease, the rigorous application of these assays will remain central to validating targets, screening for modulators, and guiding the development of novel therapeutic strategies aimed at restoring proteostasis.
Molecular chaperones, particularly Hsp70 and Hsp60, serve as critical hubs in the cellular protein homeostasis (proteostasis) network, assisting in the folding of nascent polypeptides, preventing protein aggregation, and directing the degradation of misfolded proteins [17] [6]. Their function is essential for cell survival, especially under stress conditions. However, the dysregulation of these chaperones is implicated in a range of diseases, including cancer, neurodegenerative disorders, and inflammatory conditions [17] [57] [58]. This association has driven intensive research into developing small-molecule modulators of chaperone activity. Targeting the conserved ATP-binding sites and more specialized allosteric pockets of Hsp70 and Hsp60 represents a promising therapeutic strategy. This technical guide examines the current understanding of chaperone mechanisms, explores the challenges in inhibitor development, and details the experimental approaches for discovering and characterizing compounds that target these key regulatory sites.
Structural and Mechanistic Foundations of Hsp70 and Hsp60
The Hsp70 Chaperone System
Hsp70 is a highly conserved molecular chaperone that functions as a central node in the proteostasis network. It is composed of two primary domains: an N-terminal Nucleotide-Binding Domain (NBD) that exhibits ATPase activity, and a C-terminal Substrate-Binding Domain (SBD) that interacts with client proteins [59] [17]. The SBD is further subdivided into a β-sandwich domain (βSBD) that contains the substrate-binding cleft and an α-helical "lid" domain (αSBD) that regulates affinity for clients [59].
The chaperone activity of Hsp70 is governed by an allosteric mechanism that couples nucleotide status in the NBD to substrate affinity in the SBD [59]. In the ATP-bound state, the NBD and SBD are docked, the αSBD lid is open, and the SBD has low affinity for substrates, facilitating rapid substrate binding and release. ATP hydrolysis to ADP triggers undocking of the domains, closure of the lid, and a high-affinity state for substrate binding [59] [60]. This cycle is powerfully regulated by co-chaperones: J-proteins (Hsp40s) stimulate ATP hydrolysis, while Nucleotide Exchange Factors (NEFs), such as proteins from the BAG family, promote ADP release and reset the cycle [59] [61]. This orchestrated cycle allows Hsp70 to assist in de novo protein folding, prevent aggregation, and direct clients to downstream systems like Hsp90 [60].
Table 1: Key Domains and Co-chaperones of the Hsp70 System
| Component | Structure/Features | Function in Hsp70 Cycle |
|---|---|---|
| Nucleotide-Binding Domain (NBD) | Two lobes (I & II) split into four subdomains (IA, IIA, IB, IIB); binds ATP/ADP [59]. | ATP hydrolysis drives conformational changes that allosterically regulate substrate binding affinity. |
| Substrate-Binding Domain (SBD) | Comprises βSBD (substrate-binding cleft) and αSBD ("lid" region) [59]. | Binds hydrophobic segments of client proteins; lid opening/closing modulates client affinity and release. |
| J-proteins (Hsp40s) | Contain a conserved J-domain; >45 types in humans [59] [60]. | Stimulates Hsp70's ATPase activity, coupling substrate recognition to the ATPase cycle. |
| Nucleotide Exchange Factors (NEFs) | Diverse family including BAG proteins, Hsp105, and Hsp110 [59] [60]. | Binds to the NBD to catalyze the release of ADP, allowing ATP rebinding and cycle continuation. |
Figure 1: The Hsp70 Allosteric Cycle. The chaperone alternates between low-affinity (ATP-bound) and high-affinity (ADP-bound) states for substrates, regulated by J-proteins and NEFs [59] [60].
The Hsp60 Chaperonin System
Hsp60, also known as chaperonin 60 (Cpn60), functions as a Group I chaperonin and is structurally and mechanistically distinct from Hsp70 [57] [62]. Its primary role in humans is within the mitochondria, where it facilitates the folding of newly imported proteins and refolds misfolded proteins. Hsp60 assembles into a large oligomeric complex consisting of two stacked heptameric rings, forming a central cavity that provides an isolated environment for protein folding [62].
The functional unit is the Hsp60-Hsp10 complex, where Hsp10 (the co-chaperonin, equivalent to bacterial GroES) acts as a "lid" for the folding chamber [57]. The folding cycle is driven by ATP binding and hydrolysis. An unfolded substrate protein binds to the hydrophobic apical domains of the Hsp60 ring. Upon ATP binding and Hsp10 association, the cavity undergoes dramatic conformational changes, encapsulating the substrate and promoting its folding in an isolated environment. After ATP hydrolysis, the lid disassembles, and the folded protein is released [62]. Unlike the Hsp70 system, which interacts with linear hydrophobic peptides, Hsp60 is capable of folding entire protein domains within its central cavity.
Table 2: Core Components of the Hsp60 Chaperonin System
| Component | Structure/Features | Function |
|---|---|---|
| Hsp60 (Cpn60) | Double-ring structure with 14 subunits (two heptameric rings); each subunit has apical, intermediate, and equatorial domains [62]. | Forms the central folding chamber; binds unfolded substrates and ATP; undergoes large conformational changes. |
| Hsp10 (Cpn10) | Single heptameric ring structure [57] [62]. | Functions as a detachable lid for the Hsp60 chamber, promoting substrate encapsulation. |
| Apical Domain | Located at the opening of the Hsp60 ring; contains hydrophobic residues [62]. | Serves as the primary binding site for unfolded substrate proteins and the co-chaperonin Hsp10. |
| Equatorial Domain | Forms the base of the ring; contains the ATP-binding site [62]. | Provides the structural foundation for the complex and drives ATP hydrolysis-driven conformational changes. |
Figure 2: The Hsp60 Chaperonin Folding Cycle. Unfolded proteins are encapsulated in the central cavity and fold in an isolated environment, driven by ATP and the co-chaperonin Hsp10 [57] [62].
Challenges in Targeting Chaperone ATP-Binding Pockets
Hsp70 Targeting Challenges
The development of small-molecule inhibitors against the Hsp70 ATP-binding pocket has proven exceptionally challenging for several reasons. Firstly, the ATP-binding site is flexible and hydrophilic, making it difficult to design drug-like small molecules with high affinity and suitable pharmacokinetic properties [60] [63]. Secondly, the pocket has a high affinity for endogenous nucleotides (ATP and ADP), which means potential inhibitors must compete against these high-concentration native ligands [63]. Furthermore, the functional redundancy among the multiple Hsp70 isoforms and the essential nature of Hsp70 for general cellular viability raise concerns about selectivity and toxicity for pan-Hsp70 inhibitors [60].
Hsp60 Targeting Challenges
Targeting the human Hsp60 ATP-binding pocket also presents unique hurdles. A significant challenge is achieving selectivity over bacterial homologs like GroEL, given the high degree of structural conservation, which is crucial to avoid disrupting the human microbiome [60] [57]. Additionally, because the primary function of Hsp60 is in the mitochondria, a major obstacle is the mitochondrial delivery of inhibitors; molecules must be designed to traverse both the plasma and mitochondrial membranes [60]. Moreover, a detailed understanding of the human Hsp60-Hsp10 complex's structure and regulation is still emerging, creating gaps in the structure-based design rationale [62].
Strategic Targeting of Allosteric Sites and Protein-Protein Interactions
To overcome the limitations of ATP-competitive inhibition, research has expanded to target alternative sites, a strategy that can offer improved selectivity and reduced toxicity.
Allosteric Inhibition of Hsp70
A promising approach for Hsp70 inhibition is targeting allosteric sites that disrupt the inter-domain communication essential for its function. A key breakthrough was the discovery of a cryptic secondary binding site in the NBD, adjacent to the ATP-binding pocket [63]. This site is not apparent in all Hsp70 structures but can be induced or stabilized by fragment binding. Compounds binding here can act as allosteric ATP-competitors with potentially differentiated physicochemical properties [63].
Another strategy focuses on disrupting the Hsp70-BAG protein-protein interaction. The compound JG-98, a derivative of MKT-077, binds to an allosteric site on Hsp70 and inhibits its interaction with BAG family NEFs, thereby stalling the chaperone cycle [59] [60]. This approach leverages the greater diversity of protein-protein interaction interfaces compared to deep ATP pockets, offering a path toward higher selectivity.
Allosteric Modulation of Hsp60
While the ATP-binding site of Hsp60 is also conserved, its complex oligomeric structure and interaction with Hsp10 present surfaces that can be targeted allosterically. Several natural products have been identified as Hsp60 inhibitors, though their exact mechanisms are often not fully elucidated. For instance, mizoribine (an immunosuppressant) and epolactaene have been reported to inhibit Hsp60 function, potentially by binding to sites that disrupt the conformational changes necessary for the folding cycle, rather than competing directly with ATP [57]. The structural complexity of the double-ring assembly provides multiple potential target sites for allosteric modulators that could interfere with ring assembly, co-chaperone binding, or the conformational transitions driven by ATP hydrolysis [62].
Table 3: Representative Small-Molecule Inhibitors of Hsp70 and Hsp60
| Compound | Target | Binding Site | Reported Mechanism of Action | Chemical Class |
|---|---|---|---|---|
| VER-155008 | Hsp70 | N-terminal ATP-binding pocket [59] [60]. | ATP-competitive inhibitor; blocks nucleotide binding and hydrolysis. | Adenosine-derived analog |
| MKT-077/JG-98 | Hsp70 | Allosteric site near the NBD [59] [60]. | Disrupts interaction with NEFs (e.g., BAG family); allosteric inhibitor. | Rhodocyanine derivative |
| Apoptosis Inducer | Hsp70 | N-terminal ATP-binding pocket [60]. | Inhibits Hsp70 ATPase activity. | Azole derivative |
| Mizoribine | Hsp60 | Not fully characterized [57]. | Reported to inhibit human Hsp60; used as an immunosuppressant. | Nucleoside analog |
| Epolactaene | Hsp60 | Not fully characterized [57]. | Natural product that binds to and inhibits Hsp60. | Natural product |
| Myrtucommulone A | Hsp60 | Not fully characterized [57]. | Natural product inhibitor of Hsp60. | Natural product |
Experimental Protocols for Inhibitor Discovery and Validation
Biochemical Assays for Target Engagement
ATPase Activity Assay:
- Objective: To measure the effect of a compound on the ATP hydrolysis activity of Hsp70 or Hsp60.
- Protocol: A standard coupled enzyme assay is often used. The chaperone is incubated with ATP and Mg²⁺ in the presence or absence of the test compound. The reaction mix includes phosphoenolpyruvate, pyruvate kinase, and lactate dehydrogenase. ATP hydrolysis to ADP is coupled to the oxidation of NADH, which is monitored by a decrease in absorbance at 340 nm. For Hsp70, this assay should be performed both in the presence and absence of its cognate J-protein (Hsp40) to assess the compound's impact on stimulated ATPase activity [59].
Surface Plasmon Resonance (SPR):
- Objective: To quantitatively characterize the binding affinity (KD), kinetics (kon, koff), and stoichiometry of small molecule-chaperone interactions.
- Protocol: The purified chaperone (e.g., Hsp70 NBD) is immobilized on a sensor chip. Increasing concentrations of the small molecule analyte are flowed over the surface. The association and dissociation phases of the binding event are monitored in real-time. This technique is particularly powerful for fragment-based screening, as it can detect weak binders and was instrumental in discovering fragments that bind to Hsp70's cryptic pocket [63]. Competition SPR experiments with ADP/ATP can determine if the compound is ATP-competitive.
Structural Characterization of Inhibitor Binding
X-ray Crystallography:
- Objective: To determine the high-resolution three-dimensional structure of the chaperone-inhibitor complex, revealing the precise binding site and molecular interactions.
- Protocol: The purified chaperone protein (or relevant domain) is co-crystallized with the bound inhibitor. X-ray diffraction data is collected, and the structure is solved. This method definitively identified the cryptic allosteric site in Hsp70's NBD adjacent to the ATP-binding pocket, revealing how fragments and larger compounds induce and stabilize this site [63].
Ligand-Observed Nuclear Magnetic Resonance (NMR):
- Objective: To confirm binding of a small molecule to the target protein and, in some cases, map the binding epitope.
- Protocol: Techniques such as Saturation Transfer Difference (STD) NMR are used. The protein is saturated, and magnetization is transferred to the bound ligand. Upon dissociation, the transferred magnetization is detected on the free ligand, confirming binding. This method is highly sensitive and can be used to validate hits from SPR or other screens [63].
Cellular and Functional Assays
Client Protein Degradation or Stabilization Assays:
- Objective: To assess the functional consequences of chaperone inhibition in a cellular context.
- Protocol: For Hsp70 and Hsp90 clients (e.g., oncogenic kinases, steroid hormone receptors), cells are treated with the inhibitor. The levels of the client protein are monitored via western blotting. Successful inhibition of the chaperone often leads to the proteasomal degradation of its client proteins [60].
Cell Viability and Proliferation Assays:
- Objective: To determine the cytotoxic effects and anti-proliferative potential of chaperone inhibitors, especially in cancer cell lines.
- Protocol: Assays such as the MTT or CellTiter-Glo are performed. Cells are treated with a dose range of the inhibitor, and cell viability or ATP content (a proxy for metabolically active cells) is measured after a set time (e.g., 72 hours). IC50 values are calculated to quantify potency [60].
Thermal Shift Assay (CETSA):
- Objective: To evaluate target engagement of a small molecule within the complex cellular environment.
- Protocol: Cells are treated with the compound or vehicle control, heated to different temperatures to denature proteins, and then lysed. The soluble (folded) fraction of the target chaperone is quantified by western blot. A positive shift in the protein's melting temperature (Tm) in the presence of the compound indicates stabilization and direct binding [60].
Figure 3: A Workflow for Chaperone Inhibitor Discovery and Validation. This multi-step process integrates biophysical, biochemical, and cellular methods to identify and characterize potent and specific inhibitors [60] [63].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Reagents for Investigating Hsp70/Hsp60 Biology and Inhibition
| Research Reagent | Function/Application | Example Use-Case |
|---|---|---|
| Recombinant Hsp70/Hsp60 Proteins | Purified, full-length or domain-specific proteins for in vitro assays. | Used in ATPase activity assays, SPR binding studies, and X-ray crystallography. |
| Co-chaperone Proteins (Hsp40, BAG, Hsp10) | Essential regulatory components of the chaperone cycles. | Required to study stimulated ATPase activity (Hsp40) or nucleotide exchange (BAG) in biochemical assays. |
| VER-155008 | Well-characterized, ATP-competitive Hsp70 inhibitor. | Used as a positive control in Hsp70 ATPase and cell viability assays; a benchmark compound. |
| MKT-077/JG-98 | Tool compounds that allosterically inhibit Hsp70. | Used to study the biological effects of disrupting Hsp70-NEF interactions in cellular models. |
| Epolactaene/Myrtucommulone A | Natural product Hsp60 inhibitors. | Tool compounds for probing Hsp60 function in cells and validating Hsp60 as a target. |
| ATP/ADP Analogs | Non-hydrolyzable ATP analogs or fluorescent ADP derivatives. | Used in competition binding experiments (SPR) or to monitor nucleotide binding and release. |
| Anti-Hsp70 / Anti-Hsp60 Antibodies | For detecting protein expression, localization, and stability. | Essential for western blotting, immunofluorescence, and Cellular Thermal Shift Assays (CETSA). |
The development of small-molecule inhibitors targeting Hsp70 and Hsp60 has progressed from a primary focus on competitive ATP-binding site blockade to a more nuanced strategy that includes targeting allosteric pockets and protein-protein interfaces. While significant challenges remain—including isoform selectivity, mitochondrial delivery for Hsp60, and achieving drug-like properties—recent advances are illuminating the path forward. The discovery of cryptic allosteric sites in Hsp70 and the continued exploration of the Hsp60-Hsp10 complex offer new avenues for therapeutic intervention [63] [62].
Future efforts will likely leverage integrative structural biology, including cryo-electron microscopy, to visualize full-length chaperone complexes with bound inhibitors. Furthermore, the development of bivalent molecules or PROTACs (Proteolysis-Targeting Chimeras) that simultaneously engage the chaperone and a E3 ubiquitin ligase could open new modalities for targeted protein degradation. As the understanding of chaperone biology in disease deepens, and chemical probe toolkits expand, the rational design of small-molecule modulators for Hsp70 and Hsp60 will continue to be a vibrant and critical frontier in chemical biology and drug discovery.
Disrupting Protein-Protein Interactions with Peptidomimetics and Stabilizers
Protein-protein interactions (PPIs) are fundamental mediators of virtually all biological processes, including the critical maintenance of cellular proteostasis facilitated by molecular chaperones such as Hsp70 and Hsp60 [64] [17]. The human interactome is estimated to contain between 130,000 to 650,000 PPIs, a significant portion of which remains underexplored for therapeutic intervention [64] [65]. For decades, PPIs were considered "undruggable" due to their large, flat, and relatively featureless interaction interfaces, which typically span 1,500 to 3,000 Ų, compared to the 300-1,000 Ų interfaces typically targeted by small molecules [64] [65]. However, advanced structural and chemical biological approaches have revealed that the binding energy of PPIs is often dominated by a small number of crucial amino acid residues known as "hot spots" [64] [66]. These hot spots, frequently enriched with arginine, tryptophan, and tyrosine, are defined as residues whose mutation to alanine causes a binding energy difference greater than 2.0 kcal/mol [64] [65]. This discovery has enabled the strategic development of peptidomimetics and stabilizers to selectively modulate PPIs, offering a powerful approach to intervene in disease-relevant pathways, including those regulated by the Hsp70 and Hsp60 chaperone systems in proteostasis [66] [6].
The Chaperone Framework: Hsp70 and Hsp60 in Proteostasis
Molecular chaperones form an essential machinery that maintains a healthy proteome by controlling the folding, activation, and quality control of a vast repertoire of client proteins [67] [17]. Hsp70 and Hsp60 families are central components of this cellular defense system against proteotoxic stress.
Structural and Functional Mechanisms of Hsp70
The Hsp70 family proteins, including constitutive Hsc70 and stress-induced isoforms, are conserved molecular chaperones ubiquitously expressed from prokaryotes to humans [17]. They function in multiple cellular compartments—cytosol, nucleus, endoplasmic reticulum, and mitochondria—to maintain dynamic balance in protein synthesis, folding, degradation, and translocation [17]. Structurally, HSP70 proteins share a conserved architecture comprising an N-terminal nucleotide-binding domain (NTD) that binds ATP, a C-terminal substrate-binding domain (SBD) that captures client proteins, and a flexible linker connecting these domains [17]. The SBD consists of two subdomains, SBDα and SBDβ, followed by a C-terminal tail with an EEVD motif (Glu-Glu-Val-Asp) that recognizes several co-chaperones [17].
The chaperone activity of Hsp70 operates through an allosteric conformational cycle driven by ATP hydrolysis and regulated by co-chaperones, particularly HSP40s (DnaJ proteins) [17] [68]. HSP40 proteins stimulate Hsp70's ATPase activity, enhancing its ability to bind client proteins. When bound to ADP, Hsp70 maintains a high-affinity state for unfolded substrates, crowding around them to prevent aggregation and facilitate proper folding [68]. ATP binding induces a conformational change to a low-affinity state, promoting substrate release [68]. This cycle allows Hsp70 to prevent protein aggregation, assist in refolding metastable proteins, and triage proteins for degradation [17].
Structural and Functional Mechanisms of Hsp60/Chaperonins
The Hsp60 family, known as chaperonins, facilitates protein folding through a distinctive double-ring structure that forms an enclosed folding chamber [17] [68]. In eukaryotes, Hsp60 functions primarily in mitochondria, while the TRiC/CCT complex operates in the cytosol [17]. The best-characterized system is the bacterial homolog GroEL/GroES, which forms a large complex of approximately 1 MDa [68]. GroEL (Hsp60) is a double-ring tetradecamer (14mer) with a hydrophobic patch at its opening, while GroES (Hsp10) is a single-ring heptamer that binds to GroEL in the presence of ATP or ADP [68]. This complex functions as a specialized "foldase" that encapsulates substrates within its central cavity, providing a secluded environment for proper folding without the risk of aggregation [68]. The chamber is sufficiently large to accommodate the native folding of 54-kDa GFP within its lumen [68].
Table 1: Key Chaperone Families in Proteostasis
| Chaperone Family | Major Members & Location | Structural Features | Primary Functions |
|---|---|---|---|
| HSP70 | HSPA1A, HSPA8 (Cytosol, Nucleus); HSPA5 (ER); HSPA9 (Mitochondria) [17] | Conserved NBD-SBD domains with C-terminal EEVD motif [17] | ATP-dependent foldase and holdase; prevents aggregation; triages protein fates [17] [68] |
| HSP60/Chaperonins | HSP60 (Mitochondria); TRiC (Cytosol) [17] | Double-ring structure (e.g., two heptameric rings in GroEL) [17] [68] | ATP-dependent protein foldase; prevents aggregation; encapsulates substrates [17] [68] |
| HSP40/DNAJ | DNAJA, DNAJB, DNAJC (Cytosol, Mitochondria, Nucleus) [17] | J-domain containing proteins [17] | Regulates HSP70 ATPase activity; recruits HSP70 to substrates [17] [68] |
| HSP90 | HSP90AA, HSP90AB (Cytosol); GRP94 (ER); TRAP1 (Mitochondria) [6] | Homodimer undergoing allosteric conformation changes [6] | ATP-dependent foldase for kinases, steroid receptors [17] [6] |
| Small HSPs | HSPB1-HSPB10 (Cytosol, Mitochondria, Nucleus) [6] | Large heterogeneous oligomers; α-crystallin domain [6] | ATP-independent holdase; prevents aggregation; sequesters misfolded proteins [17] [6] |
Integrated Chaperone Cycles in Proteostasis
Hsp70 and Hsp60 often function in sequential, coordinated pathways within the proteostasis network. Early-acting Hsp70 binds to nascent polypeptide chains and misfolded proteins, preventing aggregation [67] [69]. In mitochondria and bacteria, Hsp70 collaborates with the Hsp60/Hsp10 (GroEL/GroES) system, where Hsp70 can transfer partially folded clients to Hsp60 for final folding within its isolated chamber [69] [68]. The Hsp90 chaperone system further processes specific clients, particularly kinases and steroid receptors, with Hsp70 often acting upstream in the pathway [67] [6]. These chaperone cycles are regulated by nucleotide hydrolysis and coordinated by various co-chaperones that determine client protein fate—folding, activation, or degradation [67] [6].
Diagram 1: The chaperone-mediated protein folding cycle. Hsp70 initially binds nascent or misfolded polypeptides to prevent aggregation. Clients can then be transferred to Hsp60 for encapsulation and folding. Successfully folded proteins are released, while terminally misfolded proteins are targeted for degradation [67] [69] [68].
Peptidomimetic Strategies for PPI Disruption
Peptidomimetics represent a powerful class of compounds designed to mimic the essential structural and functional features of natural peptides or proteins that mediate PPIs, while overcoming the inherent limitations of native peptides, such as proteolytic susceptibility, poor bioavailability, and conformational flexibility [64] [66]. These compounds are strategically engineered to target PPI hot spots, enabling modulation of previously intractable targets.
Classification and Design Principles
Peptidomimetics are traditionally categorized into three types, recently refined into four classes (A-D) based on their similarity to the parent peptide [64] [66]:
Class A Mimetics: These are peptides with minimal alterations to side chains and backbone, closely aligning with the bioactive conformation of the parent peptide. Examples include stabilized peptides with isolated unnatural amino acids or minor backbone modifications [64] [66].
Class B Mimetics: While still peptidic, these incorporate more dramatic backbone and side chain alterations, such as peptoids, β-peptides, α/β-mixed peptides, and other foldamers that significantly diverge from natural peptide structure while maintaining key functional groups [64] [66].
Class C Mimetics: These non-peptidic molecules use synthetic scaffolds to project substituents that mimic peptide side chains. Examples include terphenyls, terephthalamides, benzoylureas, and triazine-piperazine-triazine scaffolds that spatially arrange pharmacophores to match key interaction motifs on α-helices or other secondary structures [64] [66].
Class D Mimetics: These are fully non-peptidic small molecules that mimic the mode of action without direct structural link to peptide side chains. Successful examples include ABT-737 and ABT-263 (Bcl-xL/Bak inhibitors) and Nutlin compounds (p53/MDM2 inhibitors) [64].
Table 2: Peptidomimetic Classes and Characteristics
| Class | Structural Features | Advantages | Examples |
|---|---|---|---|
| Class A | Minimal alterations to peptide sequence; natural backbone with selective modifications [64] [66] | Maintains high affinity and specificity; relatively straightforward design [66] | Peptides with D-amino acids, N-methylation, or unnatural amino acids [64] |
| Class B | Dramatic backbone alterations (e.g., β-peptides, peptoids) [64] [66] | Enhanced metabolic stability; reduced flexibility; maintained side-chain functionality [64] | β-peptides, α/β-mixed peptides, peptoids [64] |
| Class C | Non-peptidic scaffold projecting side-chain mimics [64] [66] | Improved drug-like properties; reduced peptide character; proteolytic resistance [64] | Terphenyls, terephthalamides, triazine-piperazine scaffolds [64] [66] |
| Class D | Fully non-peptidic small molecules [64] [66] | Optimal pharmacokinetics; oral bioavailability; penetration of membranes [64] | ABT-737, Nutlins, benzodiazapinediones [64] |
Secondary Structure Mimicry Strategies
Protein secondary structures, particularly α-helices, mediate a substantial proportion of therapeutically relevant PPIs, with approximately 60% of helical interfaces binding to one face of the helix [64]. Consequently, extensive research has focused on developing mimetics that replicate these structural elements.
α-Helix Mimicry: The α-helix represents the most frequently targeted secondary structure for PPI inhibition. Helix mimetics are categorized as either topographical helix mimics or stabilized helices [66]. Topographical mimics employ non-peptidic scaffolds such as terphenyls, oligoamides, or purine derivatives to project side-chain functionality vectors matching the i, i+3, i+4, and i+7 residues of an α-helix [64] [66]. Stabilized helices (foldamers) utilize chemical constraints to stabilize native peptide helices, including hydrocarbon stapling, lactam bridges, hydrogen bond surrogates, and triazole linkages [64] [66]. These constraints reduce conformational entropy, enhance proteolytic resistance, and improve cell permeability while maintaining biological activity.
Multi-Face Helix Mimicry: While early helix mimetics primarily targeted single helical faces, recent advances have yielded compounds mimicking two or three faces of the helix. Dual-faced amphipathic α-helix mimetics based on benzoylurea or triazine-piperazine-triazine scaffolds have demonstrated improved binding affinity to targets like Mcl-1 [64] [66]. Similarly, hydrogen bond surrogate helices and foldamers incorporating α- and β-amino acids can effectively mimic three faces of a helix [66].
β-Sheet and β-Strand Mimicry: Although less developed than helix mimetics, β-strand and β-sheet mimetics represent an emerging area. The design challenge lies in appropriately placing hydrogen-bonding groups while preventing self-aggregation [66]. Analysis of β-strands at PPI interfaces reveals they interact with protein partners as lone strands or sheets, with varying engagement of backbone hydrogen bonding [66]. Several scaffolds have been designed to mimic these structural features, though this area remains less explored than α-helix mimicry [66].
Experimental Methodologies for PPI Modulation
Discovery Approaches for PPI Inhibitors
Identifying and characterizing PPI modulators requires specialized methodologies beyond traditional drug discovery approaches.
High-Throughput Screening (HTS): Conventional HTS of large compound libraries has successfully identified PPI inhibitors, particularly against targets with well-defined hot spots, such as MDM2/p53 [65]. However, standard chemical libraries may not be optimal for flat PPI interfaces, necessitating libraries with greater structural diversity [65].
Fragment-Based Drug Discovery (FBDD): FBDD identifies low molecular weight fragments (200-300 Da) that bind to distinct regions of the PPI interface. These fragments are subsequently optimized through linking, growing, or merging strategies [65]. Biophysical techniques including surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), X-ray crystallography, and mass spectrometry (MS) are employed to detect and validate fragment binding [65]. FBDD has successfully yielded PPI modulators for targets including XIAP/caspase-9, Bcl-2/Bax, and bromodomains [65].
Phage and Yeast Display: Display technologies enable screening of vast peptide libraries (10^8-10^10 diversity) to identify sequences that bind protein targets [70]. In phage display, random peptide libraries are displayed on phage coat proteins, panned against immobilized targets, and enriched through multiple binding-selection-amplification cycles [70]. This approach has been instrumental in characterizing PPI interfaces and identifying inhibitory peptides for targets such as p300/HIF-1α and WWOX/ITCH [70]. Yeast, bacterial, and mammalian cell display systems offer alternative platforms, each with distinct advantages for specific applications [70].
Virtual Screening: Computational approaches screen virtual compound libraries against structural models of PPI interfaces. Structure-based virtual screening utilizes protein structural information to identify complementary compounds, while ligand-based screening employs pharmacophore models derived from known binders [65]. Virtual screening has successfully identified PPI modulators for targets including Ubc13/Uev1, MDM2/p53, and TCF/β-catenin [65].
Diagram 2: Integrated workflow for discovering PPI modulators. Following target identification, multiple screening approaches can be employed in parallel or sequence. Successful hits progress through optimization and validation stages [70] [65].
Rational Design of Selective Peptide Inhibitors
Rational design approaches leverage structural and sequence information to develop selective PPI inhibitors. A notable methodology involves identifying short homologous domains between interacting proteins [70]. For example, in developing selective inhibitors for δPKC substrate interactions, researchers aligned δPKC with its substrate PDK to identify conserved sequences, verified evolutionary conservation to confirm functional importance, and assessed sequence uniqueness to minimize off-target effects [70]. This approach yielded ψPDK, a peptide that selectively inhibits δPKC-mediated phosphorylation of PDK (K_d ~50 nM) without affecting other δPKC substrates, demonstrating remarkable specificity in both in vitro and in vivo models [70].
Stabilization Techniques for Peptide Therapeutics
Native peptides typically exhibit poor pharmacological properties due to conformational flexibility and proteolytic susceptibility. Multiple stabilization strategies have been developed to overcome these limitations:
Cyclization and Stapling: Macrocyclization and hydrocarbon stapling constrain peptides into bioactive conformations, reducing the entropic penalty of binding and enhancing proteolytic resistance. Stapled peptides targeting PPIs such as Bcl-2/BAX and MDM2/p53 have shown significantly improved cellular permeability and in vivo efficacy [64] [66].
Backbone Modification: Incorporation of N-methylated amino acids, D-amino acids, β-amino acids, or peptoid residues alters hydrogen-bonding patterns and steric properties, conferring resistance to proteases while maintaining binding affinity [64].
Unnatural Amino Acids: Incorporation of non-proteogenic amino acids such as α-aminobutyric acid (Aib) or α,α-disubstituted amino acids promotes secondary structure formation through the Thorpe-Ingold effect and prevents recognition by proteolytic enzymes [64].
Research Reagent Solutions for PPI Studies
Table 3: Essential Research Reagents for PPI and Peptidomimetic Studies
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Chaperone Proteins | Recombinant Hsp70 (HSPA1A), Hsp60 (HSPD1), Hsp90 (HSP90AA1) [17] [6] | In vitro folding assays; client interaction studies | Define chaperone-client relationships; mechanistic studies of folding pathways [67] [17] |
| Co-chaperones & Regulators | Hsp40 (DNAJA1, DNAJB1), Hop (STIP1), p23 (PTGES3) [67] [6] | Chaperone cycle reconstitution; complex assembly studies | Regulate ATPase activity; facilitate client transfer; stabilize specific conformations [67] [6] |
| Stabilized Peptide Libraries | Stapled peptides, hydrogen bond surrogate helices, β-peptide libraries [64] [66] | Screening for PPI inhibitors; specificity profiling | Target α-helix-mediated PPIs; provide proteolytic resistance and enhanced permeability [64] [70] |
| Scaffold-Based Mimetics | Terphenyl derivatives, oligobenzamides, triazine-piperazine scaffolds [64] [66] | Structure-activity relationship studies; mimetic optimization | Mimic key hot spot residues; provide non-peptidic frameworks for drug development [64] |
| Display Libraries | Phage display peptide libraries, yeast surface display libraries [70] | Peptide inhibitor discovery; epitope mapping | Identify interacting peptide sequences; characterize binding motifs [70] |
| Chemical Crosslinkers | BS3, DSS, formaldehyde [64] | PPI interface mapping; complex stabilization | Capture transient interactions; stabilize complexes for structural analysis [64] |
| ATP Analogs | ATPγS, AMP-PNP, ADP [67] [17] | Chaperone mechanism studies; nucleotide-dependent conformation analysis | Probe ATP-dependent chaperone functions; trap specific conformational states [67] [17] |
The strategic disruption of PPIs using peptidomimetics represents a transformative approach in chemical biology and drug discovery, particularly when framed within the context of Hsp70 and Hsp60 chaperone mechanisms in proteostasis. The expanding structural understanding of chaperone-client complexes, exemplified by recent cryo-EM structures of the GR-Hsp90-Hsp70-Hop loading complex and GR-Hsp90-p23 maturation complex, provides unprecedented opportunities for rational design of targeted PPI modulators [67] [6]. As classification systems for peptidomimetics evolve and stabilization techniques advance, these compounds are increasingly capable of mimicking complex secondary structure elements that mediate chaperone-client interactions. The integration of diverse discovery methodologies—from high-throughput screening and phage display to fragment-based design and virtual screening—with detailed mechanistic knowledge of chaperone cycles creates a powerful framework for developing next-generation therapeutics targeting proteostasis in cancer, neurodegenerative diseases, and other protein misfolding disorders [6] [65]. Continued advances in structural biology, computational design, and chemical synthesis will further enhance our ability to target the intricate PPI networks that govern cellular proteostasis.
Molecular chaperones, including Hsp70 and Hsp60, are fundamental components of the cellular proteostasis network, facilitating the correct folding, assembly, and degradation of proteins under both normal and stress conditions [6] [71]. In cancer, this finely balanced system is subverted through the formation of pathological epichaperomes—stable, large-scale assemblies of chaperones, co-chaperones, and other factors that reprogram cellular interactomes to support malignant phenotypes [72] [73]. Unlike the dynamic, transient complexes formed by canonical chaperones like Hsp70 and Hsp90 during their ATP-dependent folding cycles, epichaperomes are long-lived, stable scaffolding platforms [74]. They rewire protein-protein interaction (PPI) networks, enabling cancer cells to sustain proliferative signaling, survive under stress, and resist therapeutic insults [73]. This paradigm shift redefines chaperone biology in oncology, moving beyond the view of individual chaperones assisting specific client proteins to a network-level understanding where epichaperomes mastermind the pathological rewiring of cellular circuitry. Targeting these structures offers a novel, systems-level approach to cancer therapy, aiming to dismantle the very scaffolds that sustain the disease state rather than just inhibiting single proteins [72] [75].
Core Concepts: Canonical Chaperones vs. Epichaperomes
The Canonical Chaperone System: Hsp70, Hsp60, and Proteostasis
Canonical heat shock proteins, such as Hsp70 and Hsp60, are ATP-dependent molecular chaperones that play essential roles in maintaining protein homeostasis.
- Hsp70 System: Hsp70 operates through a cycle of ATP binding and hydrolysis, which drives conformational changes in its nucleotide-binding domain (NBD) and substrate-binding domain (SBD). This cycle is critically regulated by co-chaperones like Hsp40 (which delivers client proteins and stimulates ATPase activity) and nucleotide exchange factors (which facilitate ADP release) [6] [71]. The system functions dynamically and transiently to fold nascent polypeptides, refold misfolded proteins, and direct irreparably damaged proteins for degradation.
- Hsp60 Chaperonin System: Hsp60, notably in mitochondria, forms a double-ring structure that provides an isolated chamber for protein folding. Its function is aided by the co-chaperone Hsp10. The mitochondrial Hsp70 (mtHsp70/mortalin/GRP75) collaborates with Hsp60 in complexes crucial for mitochondrial protein import and folding, influencing oxidative phosphorylation and cellular metabolism [73].
The Epichaperome: A Pathological Reprogramming
The epichaperome is not merely an aggregation of overexpressed chaperones. It is a stable, hetero-oligomeric assembly that emerges in response to chronic cellular stress and disease-specific post-translational modifications (PTMs) [72] [76] [75].
Table 1: Key Differences Between Canonical Chaperones and Epichaperomes
| Feature | Canonical Chaperones (e.g., Hsp70, Hsp90) | Epichaperomes |
|---|---|---|
| Structure | Dynamic, transient complexes that disassemble post-function [74] | Stable, long-lived hetero-oligomeric assemblies [74] |
| Core Composition | Primarily HSP90/HSC70 or GRP94 with transient co-chaperones [74] | HSP90, HSC70, GRP94 with tightly bound co-chaperones and other factors [74] [73] |
| Assembly Trigger | Normal cellular processes (e.g., folding, acute stress) [74] | Chronic stress and aberrant PTMs (e.g., phosphorylation, glycosylation) [74] [76] [75] |
| Primary Function | Folding, stabilization, and degradation of client proteins [6] [74] | Scaffolding platforms that rewire protein-protein interaction (PPI) networks [72] [74] |
| Expression & Presence | Ubiquitous in all cells; levels increase under stress [74] | Context-dependent; formed predominantly in disease settings like cancer [74] [73] |
| Client Interactions | Specific, transient interactions with individual proteins [74] | Sequesters and reprograms thousands of proteins, creating disease-specific PPI networks [74] |
| Therapeutic Targeting | Traditional inhibitors block ATPase activity, causing client degradation [6] | Disruptors (e.g., PU-H71) kinetically trap and dismantle the pathological scaffold [72] [74] |
The formation of epichaperomes is driven by PTMs—particularly phosphorylation within intrinsically disordered regions (IDRs) of chaperones like Hsp90—which act as molecular encoders, stabilizing conformations that favor stable assembly over dynamic function [76] [75]. This transforms the chaperone machinery from a folding system into a network organizer that locks the cell into a pathological state.
Diagram 1: Epichaperome formation and functional impact (14 words)
The Role of Epichaperomes in Cancer Biology
Epichaperomes contribute to malignancy by acting as central hubs that dysregulate multiple cancer hallmarks.
- Rewiring of Signaling Networks: Epichaperomes serve as scaffolding platforms that facilitate aberrant protein-protein interactions, leading to the hyperactivation of oncogenic signaling pathways such as HER2 and ER in breast cancer, independent of the traditional client-protein folding paradigm [72] [74] [73].
- Metabolic Reprogramming: In mitochondria, epichaperome complexes involving TRAP1 (the mitochondrial Hsp90 paralog) and mtHsp70 can regulate oxidative phosphorylation and shift energy production toward glycolysis (the Warburg effect), a hallmark of cancer metabolism [73].
- Enhancing Cellular Adaptability: By stabilizing a pathological interactome, epichaperomes increase the ability of cancer cells to withstand various stresses, including hypoxia, nutrient deprivation, and genotoxic damage, thereby promoting survival and therapy resistance [73] [75].
The presence of epichaperomes is a marker of cellular stress and is strongly correlated with tumor dependence on these rewired networks for survival.
Detection and Analysis: Methodological Framework
A pivotal methodology for identifying and characterizing epichaperomes is the use of native polyacrylamide gel electrophoresis (Native-PAGE) followed by immunoblotting, a technique that distinguishes stable epichaperome assemblies from dynamic canonical complexes based on their molecular weight and stability [74].
Experimental Protocol: Native-PAGE Detection of Epichaperomes
Principle: Canonical HSP90 complexes are dynamic and disassemble under the conditions of native-PAGE, appearing as a broad band around 242 kDa. In contrast, the stable, tightly-bound epichaperomes persist, migrating as high-molecular-weight (HMW) species on the gel [74].
Procedure:
- Sample Preparation: Homogenize fresh or snap-frozen tumor tissues or cell pellets in a non-denaturing, detergent-free lysis buffer to preserve native protein complexes. Maintain samples at 4°C throughout.
- Native-PAGE: Load equal amounts of protein lysate onto a native polyacrylamide gel. Do not boil or add reducing agents. Run the gel under non-denaturing conditions (typically at 4°C to minimize complex dissociation).
- Western Blotting: Transfer proteins from the native gel to a membrane. Probe the membrane with antibodies against core chaperone components, such as HSP90 or HSC70.
- Analysis: Identify epichaperome-positive samples by the presence of HMW bands above the 242 kDa marker. The abundance of these HMW species directly correlates with tumor sensitivity to epichaperome-targeting drugs like PU-H71 [74].
Table 2: Key Research Reagents for Epichaperome Studies
| Reagent / Tool | Function / Utility | Experimental Application |
|---|---|---|
| PU-H71 (Zelavespib) | Small molecule that selectively binds and kinetically traps HSP90 within epichaperomes [72] [74] | Primary tool for epichaperome disruption; used for target validation, therapy, and as a chemical probe [72] [74]. |
| Native-PAGE System | Gel electrophoresis system that separates native protein complexes by size and charge [74] | Key diagnostic tool to detect stable, high-molecular-weight epichaperome complexes in cell and tissue lysates [74]. |
| HSP90/HSC70 Antibodies | Antibodies targeting core chaperone components for immunodetection. | Used in Western blotting (after native-PAGE) and immunofluorescence to identify and localize epichaperomes [74]. |
| Chemical Probes (e.g., PU-AD) | Derivatives of PU-H71 designed for specific applications like imaging or affinity purification [75] | Enable the isolation and visualization of epichaperomes for compositional and structural studies [75]. |
Diagram 2: Epichaperome detection workflow (9 words)
Therapeutic Targeting of Epichaperomes
The discovery of epichaperomes has led to a new class of therapeutic agents known as epichaperome disruptors, which function differently from traditional HSP90 inhibitors.
The PU-H71 Paradigm: From HSP90 Inhibitor to Epichaperome Disruptor
PU-H71 was initially developed as a pan-HSP90 inhibitor. However, it was discovered that its anti-tumor efficacy does not correlate with total HSP90 levels or the expression of specific client proteins. Instead, tumor sensitivity directly correlates with the abundance of epichaperomes [72] [74]. PU-H71 selectively binds to a distinct conformation of HSP90 that is incorporated into the epichaperome scaffold. It acts as a kinetic trap, binding with high stability and leading to the disassembly of the pathological structure, thereby normalizing the PPI network and inhibiting tumor growth [72] [74]. This mechanism is fundamentally different from traditional HSP90 inhibitors, which aim to broadly inhibit the chaperone's ATPase activity and induce client protein degradation.
Clinical and Preclinical Outlook
PU-H71 (zelavespib) has advanced into clinical trials for both cancer and neurodegenerative diseases like Alzheimer's, based on its ability to reverse network dysregulation [72]. This highlights the potential of epichaperome targeting as a therapeutic strategy for complex diseases driven by PPI network dysfunction.
The epichaperome represents a fundamental repurposing of the canonical chaperone machinery in cancer. It moves the pathological mechanism beyond the folding of individual oncoproteins to the orchestration of entire dysfunctional interactomes that sustain the malignant state. This new framework, built upon the foundational biology of Hsp70 and Hsp60 systems, provides a powerful rationale for developing network-centric therapeutics. Targeting the epichaperome offers a promising strategy to dismantle the core scaffolding that supports cancer progression, potentially leading to more effective and personalized treatments for a range of malignancies.
High-Throughput Screening Platforms for Chaperone Modulator Discovery
Protein homeostasis, or proteostasis, is a fundamental cellular process that ensures proteins are correctly synthesized, folded, trafficked, and degraded. The proteostasis network—an integrated system of molecular chaperones, folding enzymes, and degradation machineries—maintains the structural and functional integrity of the proteome [46]. Disruptions to this network lead to a pathological state known as dysproteostasis, which is implicated in a growing list of human diseases, including neurodegenerative disorders, metabolic syndromes, and cancer [46]. Within this network, heat shock proteins (HSPs), particularly Hsp70 and Hsp60, function as essential molecular chaperones, preventing protein misfolding and abnormal aggregation [6].
Hsp70 Chaperone Mechanism: Hsp70 operates through an ATP-dependent cycle regulated by co-chaperones. When Hsp70 binds ATP, its substrate-binding domain opens, allowing client protein interaction. ATP hydrolysis to ADP then triggers domain closure, trapping the client protein. Co-chaperones like HSP40 (DnaJ) stimulate ATP hydrolysis, while nucleotide exchange factors (e.g., BAG-1) promote ADP release and client protein folding or release [77] [6]. This cycle enables Hsp70 to assist in de novo protein folding, refold misfolded proteins, prevent aggregation, and direct irreparably damaged proteins toward degradation [78] [77].
Hsp60 Chaperone Mechanism: Hsp60, also known as HSPD1 in humans, primarily functions in mitochondria. It forms a double-ring complex that provides an isolated chamber for protein folding. Unfolded proteins enter this chamber, and upon ATP binding and hydrolysis, the chamber's lid structure undergoes conformational changes that promote client protein folding before release [78] [6].
The discovery of small molecule modulators of these chaperones provides powerful chemical tools to investigate their biology and represents a promising therapeutic strategy for proteostasis-related diseases. High-throughput screening (HTS) has emerged as a primary approach for identifying such modulators [78].
High-Throughput Screening: Core Principles and Adaptation for Chaperones
High-throughput screening (HTS) is a method for scientific discovery that uses robotics, data processing software, liquid handling devices, and sensitive detectors to rapidly conduct millions of chemical, genetic, or pharmacological tests [79]. In drug discovery, HTS allows researchers to quickly identify active compounds, antibodies, or genes that modulate a specific biomolecular pathway, providing starting points for drug design [79].
The key labware for HTS is the microtiter plate, which typically contains 96, 384, 1536, or even 3456 wells [79]. A screening facility maintains a carefully catalogued library of stock plates, and assay plates are created by pipetting small amounts (often nanoliters) from stock plates into empty plates [79]. These are then used for experiments.
Quantitative High-Throughput Screening (qHTS)
A significant advancement in HTS technology is quantitative HTS (qHTS), which profiles large chemical libraries by generating full concentration-response curves for each compound [80] [79]. In contrast to traditional "single-shot" HTS that tests one concentration per compound, qHTS tests compounds across a range of concentrations simultaneously. This approach yields pharmacological parameters such as the half-maximal effective concentration (EC50), maximal response, and Hill coefficient (nH) for the entire library, enabling the immediate assessment of nascent structure-activity relationships (SAR) and a more reliable prioritization of hit compounds [80] [79].
Table 1: Key Parameters in qHTS Data Analysis
| Parameter | Symbol | Biological Interpretation | Importance in Hit Prioritization |
|---|---|---|---|
| Half-Maximal Effective Concentration | AC50 / EC50 | Compound potency; concentration required for half-maximal effect | Lower values indicate higher potency. |
| Maximal Response | E_max / E∞ | Compound efficacy; the greatest effect a compound can produce | Measures the intrinsic activity of the compound. |
| Hill Coefficient | h / nH | Steepness of the concentration-response curve; indicates cooperativity | Can inform on the mechanism of action. |
| Baseline Response | E0 | Measured response in the absence of compound | Serves as a reference point for calculating effects. |
The data from qHTS is typically fitted to the Hill equation (also known as the four-parameter logistic model) to derive these parameters [80]: [ Ri = E0 + \frac{(E∞ - E0)}{1 + \exp{-h[\log Ci - \log AC{50}]}} ] Where ( Ri ) is the measured response at concentration ( Ci ), ( E0 ) is the baseline response, ( E∞ ) is the maximal response, ( AC_{50} ) is the half-maximal activity concentration, and ( h ) is the Hill slope [80].
HTS Assay Platforms for Hsp70 and Hsp60 Modulator Discovery
The development of robust, miniaturized assays is critical for successful HTS campaigns. For chaperone targets like Hsp70 and Hsp60, a variety of biochemical, biophysical, and cell-based assays have been employed.
Primary Screening Assays
Table 2: Primary HTS Assay Platforms for Chaperone Modulator Discovery
| Assay Type | Detection Method | Molecular Target | Readout | Key Advantages |
|---|---|---|---|---|
| Thermal Shift Assay | Fluorescence (SYPRO Orange) | Hsp70, Hsp60 | Melting Temperature (Tm) shift | Label-free; identifies direct binders that stabilize protein [77]. |
| Fluorescence Polarization (FP) | Polarized fluorescence | Hsp70-client interactions | Anisotropy change | Homogeneous assay; ideal for studying protein-ligand and protein-protein interactions [78]. |
| AlphaScreen | Luminescent proximity | Hsp70-co-chaperone PPIs | Signal amplification | Highly sensitive; suitable for detecting weak protein-protein interactions [78]. |
| Colorimetric Method | Absorbance | Hsp60 ATPase activity | Phosphate release | Low-cost and straightforward [78]. |
| ATPase Activity Assay | Luminescence/Colorimetry | Hsp70, Hsp60 ATPase | ATP consumption rate | Functional readout of chaperone activity [78] [77]. |
Experimental Protocol: Hsp70 Thermal Shift Assay
The thermal shift assay is a powerful primary screen to identify small molecules that bind and thermostabilize chaperones [77].
- Reagent Preparation: Purified Hsp70 protein is diluted in an appropriate buffer. A stock solution of the fluorescent dye SYPRO Orange is prepared. Test compounds are typically pre-plated in assay plates.
- Assay Setup:
- In a low-volume microtiter plate (e.g., 384-well), combine:
- Purified Hsp70 (final concentration ~1-2 µM)
- SYPRO Orange dye
- Test compound or control (e.g., DMSO vehicle, ADP as a positive control)
- The final assay volume is miniaturized, often to 10 µl or less [80].
- In a low-volume microtiter plate (e.g., 384-well), combine:
- Thermal Denaturation: The plate is subjected to a controlled temperature gradient (e.g., from 25°C to 95°C) in a real-time PCR instrument or other thermal cycler.
- Data Acquisition and Analysis: As the temperature increases, the protein unfolds, exposing hydrophobic regions to which the SYPRO Orange dye binds, resulting in a fluorescence increase. The point of inflection of the fluorescence curve is the protein's melting temperature (Tm). A compound that stabilizes Hsp70 will cause a positive shift in the Tm ((\Delta)Tm) compared to the DMSO control [77].
Experimental Protocol: Hsp60 Chaperone Inhibition Assay
Epolactaene/ETB is a known covalent binder that inhibits Hsp60 chaperone activity, identified through screening efforts [78]. A colorimetric assay can be used to identify similar inhibitors.
- Reagent Preparation: Purified Hsp60 protein is diluted in ATPase assay buffer (containing Mg²⁺). The test substrate (e.g., a misfolded protein) is prepared. A phosphate detection reagent (e.g., malachite green) is prepared.
- Assay Setup:
- In a microtiter plate, combine:
- Hsp60 protein
- ATP (substrate)
- Test compound or inhibitor control
- Buffer
- Initiate the chaperone reaction by adding the client protein substrate.
- Incubate at a physiological temperature (e.g., 37°C) for a fixed time to allow the ATP-dependent folding reaction to proceed.
- In a microtiter plate, combine:
- Detection:
- Stop the reaction.
- Add the colorimetric phosphate detection reagent. The amount of inorganic phosphate (Pi) released due to Hsp60's ATPase activity is proportional to its chaperone function.
- Measure the absorbance (e.g., at 620-660 nm).
- Data Analysis: A reduction in absorbance in compound-treated wells compared to controls indicates inhibition of Hsp60's ATPase and chaperone activity [78].
Hit Validation and Secondary Assays
Primary HTS hits must be rigorously validated to confirm activity and rule out false positives arising from assay interference.
- Hit Confirmation: Initial "hits" from the primary screen are re-tested in the same assay condition, often in a dose-response format (qHTS inherently provides this) to determine IC50/EC50 values and confirm reproducibility [81] [79].
- Orthogonal Assays: A different technology is used to re-confirm the hits. For an Hsp70 stabilizer identified in a thermal shift assay, an orthogonal ATPase assay or a direct binding assay like Surface Plasmon Resonance (SPR) would be employed [77] [81].
- Counter-Screening: Specific assays are run to identify compounds that interfere with the assay read-out itself (e.g., auto-fluorescent compounds, luciferase inhibitors in reporter assays) [81].
- Cellular Assays: The most critical secondary assays test the functional consequences of chaperone modulation in a cellular context.
- For Hsp70 degradative modulators like PFD-15, follow-up cellular ubiquitination and degradation assays are performed. This involves transfecting cells with a client protein (e.g., misfolded nNOS), treating with the compound, and then monitoring client ubiquitination via immunoprecipitation or its degradation via western blotting [77].
- Cell viability assays (e.g., in cancer cell lines) can assess the functional impact of Hsp90 or Hsp70 inhibitors [78].
- Luciferase refolding assays can measure the cellular chaperone activity of Hsp70 in facilitating the reactivation of heat-denatured luciferase [77].
The Scientist's Toolkit: Essential Research Reagents and Solutions
A successful HTS campaign for chaperone modulators relies on a curated set of high-quality reagents and tools.
Table 3: Research Reagent Solutions for Chaperone HTS
| Reagent / Resource | Function and Role in HTS | Specific Examples |
|---|---|---|
| Diverse Compound Library | Source of chemical starting points for screening; diversity is key to finding novel hits. | Evotec's library (>850,000 compounds); Maybridge and ChemDiv libraries [77] [81]. |
| Purified Chaperone Proteins | The primary target for biochemical HTS assays. Requires high purity and functional activity. | Recombinant human Hsp70, Hsp90, Hsp60 [77]. |
| Fluorescent Probes | Enable detection in label-free or binding assays. | SYPRO Orange (thermal shift assay) [77]. |
| Positive Control Compounds | Benchmark for assay performance and validation. | ADP for Hsp70 thermal shift assay [77]; Geldanamycin for Hsp90 inhibition [78]. |
| Client Proteins | Substrates for functional chaperone assays. | Misfolded neuronal NO synthase (nNOS) for Hsp70 [77]; Specific unfolded proteins for Hsp60. |
| Cellular Models | For secondary phenotypic and functional validation. | Engineered cell lines expressing mutant client proteins (e.g., polyQ-AR) [77]; Patient-derived iPSCs for disease context [82]. |
High-throughput screening has proven to be an indispensable strategy for discovering novel chemical modulators of Hsp70 and Hsp60. The integration of robust primary assays like the thermal shift assay with rigorous secondary validation in cellular models has yielded valuable tool compounds, such as PFD-15 for Hsp70 and epolactaene for Hsp60 [78] [77]. These compounds not only serve as starting points for therapeutic development but also as chemical probes to decipher the complex biology of chaperones in proteostasis.
The future of HTS in this field lies in the continued development of more physiologically relevant assay systems, including the use of patient-derived induced pluripotent stem cells (iPSCs) to model diseases of proteostasis [82]. Furthermore, the adoption of advanced screening technologies such as affinity selection mass spectrometry (ASMS) and DNA-encoded library (DEL) screening will provide access to novel chemical space and difficult-to-target chaperone functions [81]. As the structural biology of chaperone-cochaperone-client complexes advances [6], structure-based virtual screening and AI/ML-driven hit prioritization will work in tandem with experimental HTS to accelerate the discovery of the next generation of chaperone modulators, ultimately contributing to new therapies for cancer, neurodegenerative disorders, and other protein misfolding diseases.
Overcoming Research and Therapeutic Development Challenges
Addressing Functional Redundancy and Isoform Selectivity in Inhibitor Design
The Hsp70 and Hsp60 chaperone families represent central pillars of cellular proteostasis, facilitating the folding, assembly, and degradation of a vast repertoire of client proteins [83] [62]. Their involvement in numerous pathological conditions, including cancer, neurodegenerative diseases, and inflammatory disorders, has established them as compelling therapeutic targets [84] [5] [57]. However, the development of effective inhibitors is significantly hampered by two intrinsic biological features: functional redundancy among multiple chaperone isoforms and the high degree of conservation in their functional domains [85] [83].
Functional redundancy ensures that the loss of one chaperone isoform can be compensated by another, thereby blunting the therapeutic effect of isoform-specific inhibition [85]. Meanwhile, the structural conservation of nucleotide-binding sites across isoforms presents a formidable obstacle for achieving selective targeting, raising the potential for off-target effects [85] [6]. This technical guide examines the molecular basis of these challenges and outlines contemporary strategic frameworks and methodologies to overcome them, enabling the rational design of next-generation chaperone inhibitors.
Structural and Functional Basis of Redundancy and Selectivity
The Hsp70 Chaperone System
The Hsp70 family in humans consists of multiple members, including the cytosolic, inducible Hsp70 (HSPA1A), the constitutive Hsc70 (HSPA8), the endoplasmic reticulum-resident BiP (HSPA5), and the mitochondrial mortalin (HSPA9) [83]. All members share a conserved domain architecture comprising an N-terminal Nucleotide-Binding Domain (NBD) that exhibits ATPase activity, and a C-terminal Substrate-Binding Domain (SBD) that interacts with client proteins [83] [18]. The functional cycle is allosterically regulated by ATP binding and hydrolysis, which controls the affinity of the SBD for substrates [83] [18]. This cycle is further tuned by cohorts of co-chaperones, such as J-domain proteins (Hsp40s) that stimulate ATP hydrolysis, and Nucleotide Exchange Factors (NEFs) like Bag proteins and Hsp110 that facilitate ADP release [83] [18].
Table 1: Key Human Hsp70 Isoforms and Their Characteristics
| Isoform | Gene | Primary Localization | Expression Pattern | Notable Functions |
|---|---|---|---|---|
| Inducible Hsp70 | HSPA1A | Cytosol/Nucleus | Stress-induced | Anti-apoptosis, Cancer cell survival [83] [84] |
| Constitutive Hsc70 | HSPA8 | Cytosol/Nucleus | Constitutive | Basal protein folding, CMA [83] [18] |
| BiP | HSPA5 | Endoplasmic Reticulum | Constitutive/Inducible | ER protein folding, UPR [83] |
| Mortalin | HSPA9 | Mitochondria | Constitutive | Mitochondrial protein import, p53 regulation [83] [86] |
The high sequence conservation, particularly within the NBD, means that traditional ATP-competitive inhibitors like VER-155008 often lack sufficient isoform selectivity [85]. Compensatory mechanisms between isoforms are a major concern; for instance, significant reduction in cancer cell viability requires the simultaneous knockdown of both the inducible Hsp70 (HSPA1A) and the constitutive Hsc70 (HSPA8), as targeting either one alone is insufficient [85].
The Hsp60 Chaperone System
Human Hsp60 (HSPD1), or chaperonin 60, primarily functions as a mitochondrial chaperone that forms a large double-ring structure, creating a central cavity for protein folding in conjunction with its co-chaperone Hsp10 [62]. This complex, analogous to the bacterial GroEL/GroES system, undergoes ATP-dependent conformational changes to encapsulate and fold client proteins [62] [57]. While historically considered a mitochondrial protein, evidence indicates Hsp60 can localize to the cytosol, cell surface, and extracellular space, participating in a wider range of functions, including immune regulation and apoptosis [57].
The oligomeric state of Hsp60 is critical to its function and may be linked to its role in pathology. Under stress conditions, the equilibrium between monomers and oligomers can shift, and this appears to influence its effects on cell survival [62] [57]. The high structural similarity of its active site to bacterial GroEL complicates the development of selective inhibitors that do not disrupt essential mitochondrial function or the gut microbiome [62] [57].
Strategic Frameworks for Overcoming Redundancy and Selectivity
Targeting Isoform-Specific Protein-Protein Interactions
A powerful strategy to circumvent the conservation of nucleotide-binding sites is to target functionally critical protein-protein interfaces (PPIs) between a specific chaperone isoform and its unique co-chaperone clients [6] [86].
- Hsp70-Bag3 Interface: The interaction between Hsp70 and the co-chaperone Bag3 is a key node in a cancer-specific pro-survival pathway. The allosteric inhibitor JG-98 binds to Hsp70 and disrupts this specific PPI. This approach exploits a cancer-vulnerable pathway without completely ablating the essential, general folding functions of Hsp70, potentially widening the therapeutic window [86].
- TPR Domain Interactions: The tetratricopeptide repeat (TPR) domains of co-chaperones like CHIP and HOP bind to the conserved EEVD motif at the extreme C-terminus of Hsp70. While the EEVD motif is conserved, the TPR domains themselves offer unique, druggable surfaces that can be targeted to disrupt specific functional partnerships, such as the Hsp70-HOP-Hsp90 cascade [83] [6].
Exploiting Allosteric Pockets and Regulatory Sites
Allosteric sites are typically less conserved than orthosteric sites (like the ATP-binding pocket), offering a structural basis for achieving selectivity.
- Allosteric Hsp70 Inhibitors: JG-98 is a prime example that binds an allosteric site on Hsp70, inducing conformational changes that selectively disrupt its interaction with Bag3, while potentially sparing interactions with other co-chaperones [86].
- Substrate-Binding Domain (SBD) Targeting: The SBD of Hsp70 is less conserved than the NBD, providing an opportunity for paralog-specific inhibitor design [85]. However, this approach is challenging due to the dynamic nature and shallow hydrophobic grooves of the SBD. The inhibitor PES (pifithrin-μ) was initially reported to bind the SBD of the inducible Hsp70, though its mode of action may involve unspecific, detergent-like effects [85].
Designing Polypharmacology and Multi-Specific Agents
Instead of pursuing absolute isoform selectivity, an alternative approach is to embrace controlled polypharmacology. This involves designing single agents or combinations that simultaneously inhibit multiple redundant chaperone nodes to create a synergistic anti-disease effect [85] [6].
- Dual Hsp70/Hsc70 Inhibition: Evidence suggests that for a robust anti-cancer effect, concurrent inhibition of both the inducible Hsp70 and constitutive Hsc70 is necessary [85]. A pan-cytosolic Hsp70 inhibitor could be designed for this purpose.
- Hsp70/Hsp90 Combination: Given the close functional collaboration between Hsp70 and Hsp90 in folding oncogenic clients, combinations of Hsp70 and Hsp90 inhibitors can show synergistic activity, preventing the compensatory mechanisms that often limit the efficacy of single-agent therapies [84] [6].
Table 2: Representative Inhibitors of Hsp70 and Hsp60 and Their Characteristics
| Inhibitor | Primary Target | Proposed Mechanism | Reported Potency | Challenge Related to Redundancy/Selectivity |
|---|---|---|---|---|
| VER-155008 | Hsp70/Hsc70 | ATP-competitive, arrests NBD in half-open state [85] | Kd: ~0.3 µM (Hsc70) [85] | Lacks isoform specificity due to conserved ATP-binding site [85] |
| JG-98 | Hsp70 Family | Allosteric; disrupts Hsp70-Bag3 PPI [86] | Affinity: < 1 µM [86] | Targets a cancer-specific function, but may inhibit multiple Hsp70 paralogs [86] |
| PES (Pifithrin-μ) | Hsp70 | Binds SBD, disrupts substrate binding (unspecific mechanism) [85] | N/A | Proposed to be Hsp70-specific over Hsc70, but mechanism is non-ideal [85] |
| Mizoribine | Hsp60 | Binds GroEL/Hsp60, inhibits ATPase activity [57] | IC50: ~9 µM (GroEL) [57] | Natural product; potential for off-target effects due to conserved Hsp60/GroEL active site [57] |
| Gold(III) Porphyrin | Hsp60 | Inhibits ATPase and refolding activity [57] | IC50: ~3.5 µM (Hsp60) [57] | Synthetic compound; selectivity over other chaperones requires further validation [57] |
Essential Experimental Protocols for Characterization
Assessing Isoform Dependency via Combinatorial Knockdown
Objective: To determine which chaperone isoforms must be inhibited simultaneously to achieve a desired phenotypic outcome (e.g., reduced cancer cell viability).
Detailed Workflow:
- siRNA Transfection: Transfert target cells with siRNAs individually and in pools targeting specific Hsp70 isoforms (e.g., HSPA1A, HSPA8, HSPA5) [85]. Use non-targeting siRNA as a negative control and siRNA against a essential gene (e.g., PLK1) as a positive control for transfection efficiency.
- Validation of Knockdown: 48-72 hours post-transfection, lyse cells in RIPA buffer. Perform Western blot analysis using isoform-specific antibodies to confirm efficient protein knockdown [85].
- Viability Assay: In parallel, seed cells in 96-well plates for viability assessment. At 72-120 hours post-transfection, measure cell viability using a standardized assay (e.g., CellTiter-Glo Luminescent Cell Viability Assay). Normalize data to non-targeting siRNA controls [85].
- Data Interpretation: Significant reduction in viability only upon combined knockdown of specific isoforms (e.g., HSPA1A and HSPA8) identifies non-redundant, essential isoform pairs and validates the requirement for a multi-targeting inhibitor strategy [85].
Profiling Inhibitor Specificity and Mechanism of Action
Objective: To characterize the binding affinity, specificity, and functional impact of a candidate inhibitor across multiple chaperone isoforms.
Detailed Workflow:
- Nucleotide Binding Assay: Use a colorimetric or radiometric assay to monitor ATPase activity. Purify the NBD of different Hsp70 isoforms. Incubate with ATP and increasing concentrations of the inhibitor. An ATP-competitive inhibitor will show a characteristic change in the enzyme's kinetic parameters (e.g., increased Km for ATP) [85].
- Surface Plasmon Resonance (SPR): Immobilize purified full-length chaperone isoforms on a sensor chip. Measure the binding kinetics (Kon, Koff, KD) of the inhibitor flowing over the chip. This directly quantifies affinity and reveals isoform selectivity based on differential KD values [85].
- Co-chaperone Disruption Assay:
- Immunoprecipitation (IP): Treat cells with the inhibitor, lyse, and immunoprecipitate the target chaperone (e.g., Hsp70) with a specific antibody. Probe the immunoprecipitate by Western blot for associated co-chaperones (e.g., Bag3, Hsp40) [86].
- Plasmid-Based Assay:
Diagram 1: Workflow for testing co-chaperone disruption. This approach can confirm whether an inhibitor like JG-98 specifically disrupts the Hsp70-Bag3 interaction without affecting Hsp70's binding to other partners like Hsp40 [86].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating Chaperone Redundancy and Inhibition
| Reagent / Tool | Function in Research | Key Considerations |
|---|---|---|
| Isoform-Specific siRNAs | To selectively knock down individual chaperone paralogs and assess functional compensation [85]. | Requires validation of knockdown efficiency and specificity via qPCR/Western. |
| Recombinant Chaperone Proteins | For in vitro binding and activity assays (e.g., SPR, ATPase assays) [85]. | Purification of full-length, functional proteins is critical; tags should not interfere with activity. |
| Isoform-Specific Antibodies | To detect and quantify protein levels in validation experiments (Western blot, IP) [85]. | Must be rigorously validated for specificity against the target isoform. |
| Validated Inhibitors (e.g., VER-155008, JG-98) | As tool compounds to probe chaperone function and as benchmarks for new inhibitors [85] [86]. | Understand their precise mechanism (orthosteric vs. allosteric) and limitations (specificity). |
| Co-chaperone Expression Plasmids | To study the effect of inhibitors on specific PPIs in cellular models [86]. | Allows for manipulation of specific pathways in a controlled manner. |
The functional redundancy and high conservation of the Hsp70 and Hsp60 chaperone families are no longer insurmountable barriers to drug development. The strategic pivot from targeting conserved catalytic sites to disrupting isoform-specific protein-protein interfaces and allosteric networks represents a paradigm shift in the field. The experimental frameworks outlined herein—combinatorial knockdowns, detailed mechanistic profiling, and the use of advanced tool compounds—provide a roadmap for characterizing and validating next-generation inhibitors. By leveraging these sophisticated approaches, researchers can design multi-specific or highly selective agents that effectively modulate chaperone function for therapeutic benefit, while minimizing compensatory mechanisms and off-target effects. This progress promises to unlock the full potential of chaperone-directed therapies in oncology and beyond.
Navigating Shallow Binding Pockets and Allosteric Regulation Complexities
Targeting molecular chaperones like Hsp70 and Hsp60 represents a promising therapeutic strategy for cancer, neurodegenerative diseases, and other protein misfolding disorders. However, the development of effective chaperone-targeting compounds faces significant challenges due to the prevalence of shallow binding pockets and complex allosteric regulation networks. These structural features defy traditional drug design paradigms that rely on deep, hydrophobic pockets for high-affinity ligand binding. Within the context of proteostasis research, understanding these complexities is essential for developing novel therapeutics that can precisely modulate chaperone function without disrupting essential cellular protein homeostasis.
The chaperone systems of Hsp70 and Hsp60 exemplify these challenges, as their biological functions depend on conformational dynamics and protein-protein interactions (PPIs) that occur across extensive, often shallow, surface interfaces. This technical guide examines the structural basis of these challenges and presents advanced methodological approaches for mapping binding sites, elucidating allosteric mechanisms, and designing targeted interventions within the Hsp70 and Hsp60 chaperone networks.
Structural Basis of Shallow Binding Pockets in Chaperone Systems
Defining Characteristics of Challenging Binding Sites
Shallow binding pockets in chaperone proteins present distinct obstacles for drug discovery. These sites are characterized by several key features that limit their druggability using conventional small molecules:
- Reduced Surface Complementarity: Unlike deep pockets that allow extensive van der Waals contacts, shallow pockets provide limited surface area for ligand interaction
- High Polarity: Surface residues often contain polar or charged side chains that favor solvent exposure over hydrophobic interactions
- Conformational Flexibility: Binding interfaces frequently involve dynamic regions that undergo conformational changes during functional cycles
- Cryptic Nature: Some functionally relevant pockets are not apparent in apo-structures and only form upon ligand binding or protein-protein interaction [87]
In the Hsp70 system, the substrate-binding domain (SBD) exemplifies these challenges. The SBD contains a hydrophobic groove approximately 5 × 7 Å that accommodates client proteins—a pocket too shallow and exposed for conventional small-molecule targeting [19]. Similarly, Hsp60 (also known as CCT or TRiC) forms a complex folding chamber that interacts with client proteins through multiple transient, shallow contacts rather than defined deep pockets [88].
Structural Features of Hsp70 and Hsp60 Relevant to Binding Site Topology
Table 1: Structural Characteristics of Major Chaperone Families
| Chaperone Family | Key Domains | Binding Site Characteristics | Client Protein Interaction Mode |
|---|---|---|---|
| Hsp70 | NBD (Nucleotide-Binding Domain), SBD (Substrate-Binding Domain) with β-sandwich and α-helical lid | Shallow hydrophobic groove (~5×7 Å) in SBD; multiple allosteric sites at domain interfaces | Extended polypeptide segments captured in hydrophobic groove |
| Hsp60/CCT/TRiC | Multi-subunit double-ring assembly with multiple substrate-binding regions | Distributed, transient binding sites across chamber interior; preference for β-hairpins | Full protein domains enclosed within folding chamber; β-strand interactions |
| Small HSPs (e.g., HSPB1) | α-crystallin domain with N/C-terminal extensions | Dynamic surfaces involved in oligomer formation and client binding | Surfactant-like interactions with partially folded clients |
| HSP90 | N-terminal, Middle, and C-terminal domains | Multiple allosterically coupled sites; C-terminal domain has shallow peptide-binding site | Client proteins recruited via co-chaperone complexes rather than direct binding |
Hsp70 exhibits a conserved domain architecture consisting of an N-terminal nucleotide-binding domain (NBD) that hydrolyzes ATP, and a C-terminal substrate-binding domain (SBD) that interacts with client proteins. The SBD is further divided into a β-sandwich subdomain that forms the primary client-binding groove and an α-helical lid that regulates client access [19]. The functional cycle of Hsp70 involves allosteric communication between these domains—ATP binding to the NBD induces conformational changes that alter substrate affinity in the SBD, while substrate binding stimulates ATP hydrolysis. This allosteric mechanism creates opportunities for intervention at multiple points in the cycle, but the relevant regulatory sites often exhibit the challenging shallow characteristics described above.
Hsp60 functions as a multi-subunit complex that forms a double-ring folding chamber. Client proteins bind to the interior surface of this chamber through multiple, distributed interactions rather than a defined binding pocket. Recent cryo-EM studies of the G protein β5 β-propeller folding trajectory within CCT revealed a complex binding landscape with preferences for specific structural motifs like β-hairpins [88]. This distributed binding mode presents exceptional challenges for traditional small-molecule intervention.
Methodological Approaches for Mapping Challenging Binding Sites
Computational Mapping Techniques
Computational fragment-based mapping methods have emerged as powerful tools for identifying and characterizing shallow binding pockets in chaperone systems. These techniques simulate the binding behavior of small molecular probes to detect protein surface regions with favorable interaction properties.
Table 2: Computational Methods for Binding Site Mapping
| Method | Underlying Principle | Advantages | Limitations | Applicability to Chaperones |
|---|---|---|---|---|
| FTMap | Exhaustive docking of molecular probes followed by clustering | Fast, comprehensive coverage; consensus sites identify hot spots | Treats protein as largely rigid; continuum solvation | Excellent for initial screening of Hsp70/60 structures |
| Mixed Solvent MD (MixMD, SILCS) | MD simulations in binary solvent mixtures | Accounts for full protein flexibility and explicit solvent | Computationally intensive; limited by simulation timescale | Suitable for studying dynamic chaperone conformations |
| KeyAlloSite | Evolutionary coupling analysis between orthosteric and allosteric sites | Identifies functionally important allosteric residues | Requires sufficient homologous sequences | Effective for conserved chaperone families |
The FTMap algorithm operates by exhaustively docking a library of small organic molecules to the protein surface, evaluating billions of potential binding positions. Probe positions are clustered based on spatial proximity, and "consensus sites" where multiple different probes cluster identify binding hot spots—regions that contribute disproportionately to binding free energy [87]. For shallow pockets, FTMap can detect whether sufficient hydrophobic or polar character exists to support ligand binding.
Mixed solvent molecular dynamics (MSMD) methods, including MixMD and Site-Identification by Ligand Competitive Saturation (SILCS), employ molecular dynamics simulations of the target protein in aqueous solutions containing organic solvents at high concentrations. These methods account for protein flexibility and the competitive nature of water and probe binding, providing a more realistic representation of binding events at shallow surfaces [87]. For dynamic chaperones like Hsp70 that undergo significant conformational changes during their functional cycle, MSMD can reveal transient pockets that might be missed by rigid docking approaches.
The KeyAlloSite method represents a specialized approach that leverages evolutionary coupling analysis to identify allosteric sites with functional significance. This method is based on the principle that residues in allosteric sites show stronger evolutionary coupling with orthosteric sites compared to non-functional surface regions. By analyzing multiple sequence alignments of chaperone families, KeyAlloSite can predict key allosteric residues even in shallow surface regions [89].
Experimental Mapping Protocols
Multiple Solvent Crystal Structures (MSCS) Protocol
The MSCS method provides experimental validation of computational mapping results and involves the following steps:
Protein Crystallization: Grow initial crystals of the target chaperone protein (e.g., Hsp70 NBD or SBD) using standard vapor diffusion methods.
Soaking in Organic Solvents: Transfer crystals to stabilization solutions containing 10-50% (v/v) organic solvents such as:
- Isopropanol: Probes hydrogen bonding capacity
- Acetonitrile: Assesses dipolar interactions
- Benzene: Evaluates aromatic and hydrophobic interactions
- Acetamide: Tests amide-binding preferences
Data Collection and Structure Determination: Collect X-ray diffraction data for each soaked crystal and solve structures using molecular replacement.
Consensus Site Identification: Superimpose all structures and identify regions where organic solvent molecules cluster consistently across multiple datasets. These consensus sites represent binding hot spots [87].
NMR-Based Fragment Screening Protocol
NMR methods provide a solution-based approach for mapping binding sites without requiring crystallization:
Isotope Labeling: Express the target chaperone domain (e.g., Hsp70 SBD) in E. coli grown in (^{15})N-enriched media to produce uniformly (^{15})N-labeled protein.
Spectra Collection: Acquire (^{1})H-(^{15})N HSQC spectra of the labeled protein in the absence and presence of fragment libraries.
Chemical Shift Perturbation Analysis: Monitor changes in backbone amide chemical shifts upon fragment addition. Residues showing significant perturbations ((> \mu + \sigma) of all changes) map binding sites.
STD-NMR for Weak Binders: For fragments with fast exchange, use saturation transfer difference NMR to confirm binding and characterize the binding epitope.
Allosteric Regulation Networks in Hsp70 and Hsp60 Chaperones
Hsp70 Allosteric Mechanisms
The Hsp70 chaperone system exemplifies sophisticated allosteric regulation that coordinates nucleotide status with substrate binding. The structural basis of this allostery involves communication between the nucleotide-binding domain (NBD) and substrate-binding domain (SBD):
Hsp70 Allosteric Cycle: This diagram illustrates the conformational cycle of Hsp70, showing the allosteric coupling between nucleotide status and substrate-binding affinity. The cycle is regulated by co-chaperones: J-domain proteins (JDPs) stimulate ATP hydrolysis, while nucleotide exchange factors (NEFs) promote ADP release and reset the cycle [19] [40].
The allosteric regulation of Hsp70 is critically modulated by co-chaperones. J-domain proteins (JDPs) interact with Hsp70 to stimulate its ATPase activity, while nucleotide exchange factors (NEFs) facilitate ADP release. The human genome encodes approximately 50 JDPs and 8 NEFs, which confer remarkable specificity to the Hsp70 system by targeting it to specific subcellular locations and client proteins [40]. This network architecture allows Hsp70 to participate in diverse cellular processes including protein folding, complex assembly, and protein degradation.
Hsp60/CCT Folding Mechanism
The Hsp60 chaperonin system, known as CCT or TRiC in eukaryotes, employs a different allosteric mechanism based on large-scale conformational changes:
CCT/TRiC Folding Cycle: This diagram illustrates the allosterically regulated folding cycle of the Hsp60 chaperonin CCT/TRiC. ATP binding induces large-scale conformational changes that enclose client proteins in a protected folding chamber, while synchronized ATP hydrolysis resets the complex for subsequent folding rounds [88] [3].
Recent cryo-EM studies have visualized the folding trajectory of specific clients like the G protein β5 β-propeller within CCT, revealing a multi-stage process with distinct folding intermediates. The chamber interior contains multiple shallow binding sites that interact with different structural elements of the client protein throughout its folding trajectory [88].
Experimental and Computational Toolkit for Chaperone Research
Research Reagent Solutions
Table 3: Essential Research Reagents for Chaperone Binding Studies
| Reagent/Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Fragment Libraries | Maybridge Ro3, DSI-Poised Library | Initial screening for shallow binding sites | Typically 500-1500 compounds with MW < 300; high aqueous solubility |
| Mapping Probes | Isopropanol, Acetonitrile, Benzene, Acetamide | MSCS experiments to identify binding hot spots | Use at 10-50% concentration in crystallization solutions |
| Isotope-Labeled Proteins | (^{15})N-Hsp70 SBD, (^{13})C(^{15})N-Hsp60 subunits | NMR studies of binding and allostery | Require specialized expression in minimal media with isotopic precursors |
| TRUPATH BRET Sensors | Gα subunit expression plasmids | Monitoring G protein activation in cellular contexts | Validated for 14 Gα proteins; requires luminescence detection |
| QconCAT Standards | Synthetic genes encoding concatenated peptide standards | Absolute quantification of chaperones and clients by SRM/MS | Designed to match proteolytic peptides from target proteins |
| Cryo-EM Reagents | Graphene oxide grids, Uranyl formate | High-resolution structure determination of chaperone-client complexes | Requires access to high-end cryo-electron microscopes |
Targeting Strategies for Challenging Chaperone Sites
Therapeutic targeting of shallow binding pockets in chaperones requires innovative approaches beyond traditional small molecules:
Beyond Rule of Five (bRo5) Compounds: These larger molecules (MW > 500) can form extensive interactions with shallow surfaces. For example, the thrombin inhibitor Argatroban extends to five hot spots in its target despite its relatively high molecular weight [87].
Protein-Protein Interaction Inhibitors: Designed to disrupt specific chaperone-co-chaperone or chaperone-client interactions. Successful examples often mimic secondary structure elements like α-helices or β-turns [87].
Allosteric Modulators: Target structurally distinct sites that regulate function at a distance. The intracellular NTSR1 modulator SBI-553 exemplifies this approach by binding at the GPCR-transducer interface and changing G protein subtype selectivity [90].
Molecular Glues: Stabilize specific PPIs rather than inhibiting them. This approach can redirect chaperone activity toward specific client proteins or functional outcomes.
For Hsp70, targeting strategies have evolved through four developmental stages: (1) pan-isoform inhibitors (1990s), (2) isoform-selective inhibitors (2000s), (3) PPI inhibitors targeting chaperone-co-chaperone interactions (2010s), and (4) multi-specific molecules that simultaneously engage multiple sites (2020s) [6]. This progression reflects the growing sophistication of approaches to overcome the challenges posed by shallow binding pockets.
The complexities of shallow binding pockets and allosteric regulation in Hsp70 and Hsp60 chaperone systems represent both significant challenges and opportunities for therapeutic intervention. Successful navigation of these complexities requires integrated approaches that combine computational mapping, structural biology, and mechanistic studies of allosteric networks. The methodological framework presented in this guide provides a pathway for identifying and characterizing functionally relevant sites in these challenging targets, even when they lack the canonical features of druggable pockets.
As proteostasis research advances, understanding the tissue-specific expression of chaperone networks [3] and their alterations in disease states will enable more precise targeting strategies. The layered architecture of core and variable chaperones across human tissues suggests that tissue-specific therapeutic interventions may be possible by targeting the variable components of the chaperone network [3]. Furthermore, the conservation of allosteric mechanisms across protein families [89] provides opportunities for leveraging insights from one system to inform targeting strategies in others.
Ultimately, mastering the complexities of shallow binding pockets and allosteric regulation in chaperone systems will expand the druggable proteome and enable novel therapeutic strategies for the many diseases characterized by proteostasis dysfunction.
Mitochondrial Delivery Challenges for Hsp60-Targeted Compounds
Heat shock protein 60 (Hsp60) is a highly conserved molecular chaperone that plays an indispensable role in cellular proteostasis, primarily within the mitochondrial matrix [1] [91]. As a Group I chaperonin, Hsp60 forms a complex folding machinery with its co-chaperone Hsp10, facilitating the correct folding of newly imported proteins and the refolding of misfolded polypeptides in an ATP-dependent manner [27] [92]. This function is critical for maintaining mitochondrial integrity, regulating oxidative stress, and preventing apoptosis [1]. In recent years, Hsp60 has emerged as a compelling therapeutic target across various pathological conditions, including cancer, inflammatory diseases, and neurodegenerative disorders [60] [1] [93]. However, the development of effective Hsp60-targeted compounds faces a formidable obstacle: the complex biological barriers that separate the compound's site of administration from its site of action within the mitochondrial matrix. This whitepaper examines these delivery challenges within the broader context of chaperone mechanisms, analyzes current experimental approaches to overcome them, and provides a strategic toolkit for researchers engaged in this demanding field of drug development.
Hsp60 Structure, Function, and Compartmentalization
Structural Organization and Chaperoning Cycle
The human Hsp60 protein is a nuclear-encoded protein synthesized in the cytosol as a precursor containing an N-terminal mitochondrial import signal (MIS) of 26 amino acids [94] [91]. Each Hsp60 monomer consists of three distinct domains: the apical domain (binds substrate and Hsp10), the intermediate domain (acts as a flexible hinge), and the equatorial domain (contains the ATP-binding site and mediates oligomerization) [92] [91]. Upon successful mitochondrial import and cleavage of its MIS, Hsp60 self-assembles into a dynamic oligomeric structure.
Unlike its bacterial homolog GroEL, which forms a stable tetradecamer, human mitochondrial Hsp60 (mtHsp60) exhibits a more complex equilibrium between different oligomeric states [94] [93]. Biophysical studies reveal that mtHsp60 exists in a concentration-dependent dynamic equilibrium between single-ring heptamers and double-ring tetradecamers [94]. The functional chaperoning cycle involves a coordinated interaction with Hsp10 and ATP hydrolysis. The cycle begins with a single-ring Hsp60 complex that binds an unfolded polypeptide substrate via hydrophobic interactions in its open cavity. Subsequently, Hsp10 and ATP bind, triggering a conformational change that encapsulates the substrate within a hydrophilic folding cage. ATP hydrolysis drives the folding process, culminating in the release of the native protein and the dissociation of the complex [27] [92] [91].
Extramitochondrial Localizations and Therapeutic Implications
While Hsp60 is predominantly mitochondrial, a growing body of evidence confirms its presence in other cellular compartments, including the cytosol, plasma membrane, and extracellular space, often in association with pathological states like cancer [94] [93]. This extramitochondrial Hsp60 can exist in its "naïve" form, retaining the MIS, and has been shown to exhibit both pro- and anti-apoptotic functions [94] [93]. The presence of Hsp60 in these non-canonical locations complicates the therapeutic landscape. Inhibitors designed to target mitochondrial Hsp60 could potentially interact with extramitochondrial pools, leading to off-target effects. Conversely, this distribution also presents opportunities for developing diagnostic biomarkers based on extracellular Hsp60 levels [93]. Therefore, a critical objective in drug design is to achieve subcellular targeting specificity to ensure compounds primarily engage the intended mitochondrial pool of Hsp60.
The Mitochondrial Delivery Challenge: A Multi-Tiered Barrier
Targeting Hsp60 requires compounds to navigate a series of sequential biological barriers. The table below summarizes the key properties of these barriers and the consequent implications for drug design.
Table 1: Multi-Tiered Biological Barriers to Mitochondrial Hsp60-Targeted Compounds
| Barrier Tier | Barrier Properties | Impact on Compound Delivery |
|---|---|---|
| 1. Plasma Membrane | Phospholipid bilayer with selective permeability; efflux pumps [60]. | Limits cellular uptake of hydrophilic or large molecules; potential active export. |
| 2. Cytosolic Environment | Crowded molecular environment (150-300 mg/mL); degradative enzymes [60]. | Promotes aggregation, non-specific binding, and enzymatic degradation during transit. |
| 3. Mitochondrial Membranes | Double-membrane system (Outer - OMM, Inner - IMM); distinct protein import machinery (e.g., TOM, TIM) [94] [95]. | The IMM presents a high electrochemical gradient (~180 mV); requires specific targeting signals for import. |
| 4. Mitochondrial Matrix | Viscous, protein-dense environment; requires correct localization for Hsp60 binding [1]. | Final hurdle for engagement with tetradecameric Hsp60-Hsp10 complex; off-target binding possible. |
The most selective barrier is the mitochondrial inner membrane (IMM). The bacterial origin of mitochondria means their import machinery is highly specialized and distinct from the nuclear import system. The translocase of the inner membrane (TIM) complex is evolutionarily designed to recognize specific mitochondrial targeting signals on native protein precursors [94] [95]. Small molecule compounds lack these endogenous signals, making their transit across the IMM a primary bottleneck. Furthermore, the matrix itself is a confined space where the functional Hsp60 complex operates, necessitating that the drug not only enter the matrix but also correctly position itself to modulate the chaperone's activity without disrupting essential mitochondrial functions, such as oxidative phosphorylation [1].
Quantitative Analysis of Hsp60 Inhibitors and Delivery Hurdles
The development of Hsp60-targeted small molecules is less advanced compared to inhibitors for other chaperones like Hsp90. The following table catalogs representative Hsp60 inhibitors and the specific delivery challenges associated with their use.
Table 2: Hsp60 Inhibitors and Associated Delivery Challenges
| Compound / Class | Reported Mechanism of Action | Documented Delivery/Mitochondrial Challenge |
|---|---|---|
| Mizoribine | Natural product; first identified Hsp60 inhibitor [60]. | Limited data on mitochondrial penetration; primarily used as an immunosuppressant. |
| Epolactaene | Natural product; binds covalently to Hsp60 [60]. | Mitochondrial delivery efficiency and specificity are not well-characterized. |
| Myrtucommulone | Natural product from myrtle; inhibits Hsp60 ATPase activity [60]. | Unknown mitochondrial uptake; potential non-specific interactions due to hydrophobic nature. |
| KSH101 | Synthetic small molecule inhibitor [60]. | Lacks a defined mitochondrial targeting strategy; efficacy may be limited by cytosolic dispersion. |
| Suvanin | Natural product from marine sponge [60]. | High molecular weight and complex structure likely impede membrane permeability. |
A critical observation from the literature is that while several Hsp60 inhibitors have been identified, their mitochondrial delivery is often assumed rather than experimentally verified [60]. The primary focus has typically been on target engagement and phenotypic efficacy in cellular models, with less emphasis on quantifying subcellular pharmacokinetics. This gap underscores the need for standardized assays to directly measure and optimize the mitochondrial accumulation of lead compounds. Furthermore, the functional redundancy within the chaperone system, where inhibition of Hsp60 can trigger compensatory upregulation of Hsp70 or other stress response pathways, adds a layer of biological complexity beyond pure compound delivery [60] [17].
Experimental Protocols for Assessing Mitochondrial Delivery and Compound Efficacy
Protocol: Mitochondrial Isolation and Subcellular Fractionation
This protocol is fundamental for directly quantifying the mitochondrial accumulation of a candidate compound.
- Cell Lysis and Homogenization: Culture relevant cell lines (e.g., HeLa, HEK293) and harvest ~10^7 cells. Wash with ice-cold PBS. Resuspend the cell pellet in isotonic mitochondrial isolation buffer (e.g., 225 mM mannitol, 75 mM sucrose, 10 mM MOPS, 1 mM EGTA, pH 7.2) supplemented with 0.1% fatty-acid-free BSA. Homogenize using a tight-fitting Dounce homogenizer (30-40 strokes) or a nitrogen cavitation chamber to efficiently break the plasma membrane while preserving mitochondrial integrity.
- Differential Centrifugation:
- Centrifuge the homogenate at 1,000 × g for 10 minutes at 4°C to pellet nuclei and unbroken cells.
- Transfer the supernatant to a fresh tube and centrifuge at 10,000 × g for 15 minutes at 4°C. The resulting pellet constitutes the crude mitochondrial fraction.
- The supernatant from this step represents the cytosolic fraction.
- Mitochondrial Wash and Purity Assessment: Gently resuspend the mitochondrial pellet in isolation buffer without BSA and repeat the high-speed centrifugation. To assess fraction purity, perform Western blotting on equal protein amounts from each fraction:
- Mitochondrial Marker: Hsp60, COX IV.
- Cytosolic Marker: Lactate Dehydrogenase (LDH), GAPDH.
- Nuclear Marker: Lamin B1.
- Compound Quantification: Lyse the purified mitochondrial and cytosolic fractions. Use Liquid Chromatography-Mass Spectrometry (LC-MS/MS) to detect and quantify the concentration of your test compound in each fraction. Compare this to a standard curve to determine the absolute mitochondrial accumulation.
Protocol: Assessing Hsp60 Functional Engagement in a Cellular Context
This protocol evaluates the functional consequences of Hsp60 inhibition, indirectly informing on successful target engagement.
- Treatment and Protein Extraction: Treat cells with the candidate compound for a predetermined time (e.g., 4-24 hours). Include vehicle control and a reference Hsp60 inhibitor (e.g., Mizoribine) as controls.
- Assessment of Mitochondrial Proteostasis (UPRmt): Lyse cells and analyze lysates by Western blot for markers of the Mitochondrial Unfolded Protein Response (UPRmt) [95], which is activated upon Hsp60 dysfunction:
- Key markers: HSP60, HSP10, ATF5.
- An increase in these markers suggests functional perturbation of the mitochondrial folding environment.
- Assessment of Apoptotic Commitment: Since Hsp60 perturbation can induce apoptosis, probe for cleavage of caspase-3 and its substrate PARP by Western blot.
- Client Protein Analysis: Identify known Hsp60 client proteins (e.g., specific mitochondrial enzymes) and monitor their levels and folding status via Western blot or native gel electrophoresis. A decrease in mature, folded clients indicates successful functional inhibition of Hsp60.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Hsp60 and Mitochondrial Delivery Studies
| Reagent / Tool | Function and Utility in Hsp60 Research |
|---|---|
| Recombinant Human Hsp60/Hsp10 Proteins | For in vitro ATPase assays, binding studies (SPR, ITC), and high-throughput screening (HTS) of compound libraries to identify direct binders/inhibitors without delivery complications [94]. |
| Hsp60-Specific Antibodies | Critical for Western blotting to assess UPRmt activation, monitor Hsp60 expression levels, and confirm subcellular localization via immunofluorescence or immunogold EM [94] [93]. |
| Mitochondrial-Targeted Fluorescent Dyes (e.g., MitoTracker) | Used in conjunction with immunofluorescence to visualize mitochondrial morphology and colocalization studies of tagged compounds or client proteins [1]. |
| Mitochondrial Isolation Kits | Commercial kits for rapid and efficient preparation of mitochondrial fractions from tissues or cultured cells for biochemical assays and compound uptake studies [94]. |
| Cellular Models with Modulated Hsp60 | Hsp60-knockdown (siRNA/shRNA) or knockout (CRISPR-Cas9) cell lines to study loss-of-function phenotypes. Cells overexpressing wild-type or mutant Hsp60 for functional rescue and mechanistic studies [1] [91]. |
| Chemical Inhibitors (e.g., Mizoribine) | Well-characterized, albeit imperfect, reference compounds for benchmarking the potency and efficacy of novel Hsp60-targeting molecules in cellular and biochemical assays [60]. |
The path to developing clinically viable Hsp60-targeted therapeutics is fraught with the formidable challenge of achieving efficient mitochondrial delivery. Current research, while identifying promising inhibitory scaffolds, often lacks rigorous validation of subcellular targeting. Future efforts must prioritize the integration of mitochondriotropic motifs, such as triphenylphosphonium (TPP+) cations or mitochondrial-penetrating peptides (MPPs), into lead compounds to actively drive accumulation across the IMM. Furthermore, the application of advanced techniques like super-resolution microscopy and cryo-electron tomography will be crucial to visualize the precise subcellular location of drug molecules and their interaction with the Hsp60-Hsp10 complex. By adopting a multi-disciplinary strategy that equally emphasizes medicinal chemistry, cell biology, and pharmacokinetics, researchers can transform Hsp60 from a compelling biological target into a tangible therapeutic reality.
Strategies for Differential Targeting of Bacterial vs. Human Chaperonin Homologs
Molecular chaperones, including Hsp70 and Hsp60 families, are essential components of cellular proteostasis networks. While these chaperones are highly conserved from bacteria to humans, their structural and functional distinctions create valuable opportunities for therapeutic intervention. Differential targeting of bacterial versus human homologs represents a promising strategy for combating infectious diseases without disrupting human protein homeostasis. This technical guide examines the mechanistic foundations and experimental approaches for selectively inhibiting bacterial chaperonins, with particular focus on DnaK (bacterial Hsp70) and GroEL (bacterial Hsp60). We explore key structural variations, allosteric regulation mechanisms, and emerging chemical biology tools that enable species-specific chaperone targeting, providing a framework for antibiotic development and proteostasis research.
The Central Role of Molecular Chaperones
Molecular chaperones constitute a complex system for client protein regulation that prevents protein misfolding and abnormal aggregation, modulates protein homeostasis, and protects cells from damage under changing environmental conditions [6]. These highly conserved proteins function as critical defense molecules against proteotoxic stress, with their abnormal expression or dysfunction being closely linked to numerous diseases including cancers, neurodegenerative disorders, and infectious diseases [6] [5].
The Hsp70 and Hsp60 families represent two major chaperone systems with distinct yet complementary functions. Hsp70 proteins operate as ATP-dependent chaperones that assist in nascent protein folding and salvage misfolded proteins, while Hsp60 chaperonins form large double-ring structures that provide an isolated environment for protein folding [96] [1]. Understanding the mechanistic differences between bacterial and human homologs of these chaperones enables the development of selective inhibitors that disrupt pathogenic proteostasis while sparing host functions.
Strategic Importance of Differential Targeting
The development of selective chaperone inhibitors represents a promising avenue for antibacterial therapy, particularly against multi-drug resistant pathogens. Bacterial DnaK and GroEL are essential for pathogen survival under stress conditions, including antibiotic exposure [97]. However, the high sequence conservation between bacterial and human chaperones (e.g., 50-70% identity for DnaK/Hsp70) presents a significant challenge for selective inhibition [97]. Successful targeting strategies must exploit subtle structural and dynamic differences to achieve specificity.
Table 1: Key Chaperonin Targets for Differential Inhibition
| Chaperone | Bacterial Homolog | Human Homolog | Sequence Identity | Essential for Pathogen Survival |
|---|---|---|---|---|
| Hsp70 | DnaK | Hsp70/Hsc70 | 50-70% | Yes [97] |
| Hsp60 | GroEL | HSP60/HSPD1 | ~50% | Yes [1] [57] |
Structural and Functional Basis for Differential Targeting
Hsp70/DnaK Architecture and Allosteric Regulation
The Hsp70 family exhibits a conserved domain architecture consisting of an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD), connected by a flexible linker that enables allosteric communication [97]. The NBD contains four subdomains (IA, IB, IIA, IIB) that form a binding cleft for ATP coordination, while the SBD comprises a β-sandwich domain for peptide binding and an α-helical lid domain [19] [96].
Hsp70 proteins function through an allosteric cycle that alternates between open (ATP-bound) and closed (ADP-bound) conformations [97]. In the open state, ATP binding to the NBD creates a low-affinity, fast-exchange state for substrates. ATP hydrolysis transitions the chaperone to a closed conformation with high substrate affinity. Nucleotide exchange factors (NEFs) then catalyze ADP-ATP exchange, resetting the cycle [19] [96]. This allosteric regulation is conserved but exhibits species-specific variations that can be exploited therapeutically.
Diagram 1: Allosteric cycle of Hsp70/DnaK chaperones. Bacterial co-chaperones DnaJ and GrpE present potential targets for selective inhibition.
Hsp60/GroEL Structural Organization and Folding Mechanism
The Hsp60 chaperonins form large double-ring structures with central folding cavities. Group I chaperonins (including bacterial GroEL and human HSP60) require a co-chaperonin lid (GroES/HSP10) for function, while Group II chaperonins (eukaryotic TRiC/CCT) contain built-in lid structures [1]. GroEL consists of two heptameric rings that form a barrel-like structure, with each subunit containing three domains: apical (substrate binding), intermediate (hinge), and equatorial (ATP binding) [1] [57].
The chaperonin folding cycle involves several coordinated steps: (1) binding of unfolded substrate to the open ring's apical domains, (2) GroES binding and ATP hydrolysis that triggers conformational changes encapsulating the substrate, (3) productive folding in the isolated chamber, and (4) release of the folded protein upon ATP hydrolysis in the opposite ring [1] [57]. The structural variations in the apical domain substrate-binding residues and the inter-ring salt bridges between bacterial and human Hsp60 create opportunities for selective targeting.
Table 2: Structural Differences Between Bacterial and Human Chaperonins
| Structural Feature | Bacterial Chaperones | Human Chaperones | Targeting Implications |
|---|---|---|---|
| Hsp70 Substrate-Binding Cavity | Hydrophobic groove with specific L1,2/L3,4 loop configurations [97] | Differing charge distribution and loop dynamics [97] | Peptide inhibitors with species-specific residue preferences |
| Hsp60 Apical Domain | Conserved hydrophobic residues for substrate binding [1] | Altered hydrophobicity patterns and charged residues [57] | Small molecules targeting species-specific substrate interactions |
| Inter-ring Interfaces | Specific salt bridges (e.g., R452-E462) [1] | Varied or absent salt bridge conservation [1] | Compounds disrupting oligomerization with species specificity |
| Allosteric Networks | Distinct nucleotide coordination and communication pathways [96] | Modified allosteric regulation and cofactor interactions [19] | Allosteric inhibitors exploiting communication differences |
Experimental Approaches for Differential Inhibition
Hot Spot Residue Identification and Molecular Dynamics
Molecular dynamics (MD) simulations enable the identification of "hot spot" residues critical for species-specific chaperone function. This approach analyzes differential conformations of bacterial and human chaperones bound to inhibitor peptides to identify residues exhibiting stable and selective interactions [97].
Experimental Protocol: Hot Spot Identification
- Structure Preparation: Obtain crystal structures of bacterial DnaK (e.g., E. coli 2KHO) and human Hsp70. Model missing regions using homology modeling.
- System Setup: Solvate chaperone structures in explicit water boxes, add ions to physiological concentration.
- Peptide Docking: Dock known inhibitor peptides (e.g., Api88, drosocin) to substrate-binding domains.
- MD Simulations: Perform extensive MD simulations (100+ ns) of chaperone-peptide complexes.
- Interaction Analysis: Calculate interaction energies, hydrogen bonding patterns, and contact frequencies.
- Conservation Mapping: Map interacting residues to sequence alignments of bacterial vs. human homologs.
- Pharmacophore Modeling: Develop 3D pharmacophore models incorporating species-specific interactions.
This methodology identified key DnaK residues (e.g., V400, F402, L414, V436 in A. baumannii) that form stable interactions with inhibitor peptides but differ in human Hsp70, enabling the design of selective inhibitors [97].
High-Throughput Screening for Allosteric Inhibitors
High-throughput screening approaches can identify compounds that allosterically inhibit chaperone networks without competing directly with ATP binding, reducing potential off-target effects [96].
Experimental Protocol: HTS for Chaperone Inhibitors
- Assay Development: Establish a luminescence-based ATPase assay detecting ADP production.
- Cofactor Optimization: Determine optimal chaperone:cofactor ratios (e.g., 10:1:1 DnaK:DnaJ2:GrpE) that maximize signal-to-noise.
- Library Screening: Screen compound libraries (e.g., ~1300 FDA-approved drugs) at ATP KM concentration.
- Hit Validation: Confirm hits using dose-response curves and exclude luciferase inhibitors.
- Mechanistic Studies: Employ cross-linking, NMR, and X-ray crystallography to determine inhibition mechanisms.
- Specificity Testing: Counter-screen against human homologs and essential enzymes.
This approach identified telaprevir, an HCV protease inhibitor, as an allosteric inhibitor of Mtb DnaK (IC50 ~4.0 μM) that mimics peptide substrates and disrupts cofactor-mediated activation [96].
Diagram 2: High-throughput screening workflow for identifying selective chaperone inhibitors.
Covalent Targeting Strategies
Covalent inhibitors can achieve enhanced selectivity by targeting non-conserved cysteine residues in chaperone structures. This approach exploits differential nucleophilicity and accessibility of cysteine thiol groups between bacterial and human homologs.
Experimental Protocol: Covalent Inhibitor Development
- Cysteine Mapping: Identify non-conserved cysteine residues in bacterial chaperones (e.g., Cys-574 and Cys-603 in human Hsp70 SBDα are not conserved in all bacterial DnaKs).
- Electrophile Design: Synthesize compounds with appropriate electrophilic warheads (e.g., Michael acceptors, α,β-unsaturated carbonyls).
- Kinetic Characterization: Determine second-order rate constants for covalent modification.
- Structural Analysis: Solve crystal structures of covalent chaperone-inhibitor complexes.
- Functional Assessment: Evaluate effects on ATPase activity, substrate binding, and cofactor interactions.
- Cellular Activity: Test membrane permeability and antibacterial efficacy.
This strategy has been successfully applied using PES (pifithrin-μ), which covalently modifies Cys-574 and Cys-603 in human Hsp70, altering SBDα structure and function [98]. Similar approaches could target non-conserved cysteines in bacterial chaperones.
Research Reagent Solutions
Table 3: Essential Research Reagents for Chaperone Targeting Studies
| Reagent/Category | Specific Examples | Function/Application | Species Specificity Considerations |
|---|---|---|---|
| Inhibitor Compounds | Telaprevir, PES (pifithrin-μ), Mizoribine, Myrtucommulone | Mechanistic probes and therapeutic leads | Telaprevir inhibits Mtb DnaK; PES targets human Hsp70 Cys residues [96] [98] |
| Peptide Inhibitors | Api88, Drosocin, Oncocin, Pyrrhocoricin | Substrate-competitive inhibitors; study SBD interactions | Proline-rich insect peptides with preferential DnaK binding [97] |
| Antibodies | Anti-HSPA6 (SPA-754), Anti-DNAJB1 (SPA-400), Anti-HSPH1 (ab109624) | Detection, localization, and functional assays | Species-specific antibodies essential for differentiating homologs [99] |
| Expression Systems | E. coli recombinant protein expression, SH-SY5Y neuronal cells | Production of chaperone proteins and cellular studies | Human-specific HSPA6 requires human cell lines [99] |
| Screening Assays | Luciferase-coupled ATPase assay, Thermal shift assay, Proteostasis stress assays | Compound screening and mechanistic evaluation | Optimize cofactor ratios for species-specific chaperone networks [96] |
The strategic targeting of bacterial chaperonins through species-specific structural and mechanistic differences represents a promising approach for antibacterial development. The integration of structural biology, computational modeling, and chemical biology has enabled significant advances in identifying selective inhibitors against both Hsp70 and Hsp60 family members. Future efforts should focus on exploiting the unique allosteric networks of bacterial chaperones and developing multi-specific compounds that simultaneously target essential chaperone-cofactor interactions. As structural insights into chaperone mechanisms continue to grow, so too will opportunities for innovative therapeutic strategies that disrupt pathogenic proteostasis while preserving host protein homeostasis.
Managing Chaperone Inhibition Toxicity and Therapeutic Window Optimization
Molecular chaperones, including Hsp70 and Hsp90, constitute an essential network responsible for maintaining protein homeostasis (proteostasis) within cells by facilitating the correct folding, assembly, and degradation of a vast repertoire of client proteins [17]. In pathological states such as cancer, this chaperone machinery is co-opted to stabilize overexpressed and mutated oncoproteins, creating a dependency that establishes chaperones as high-value therapeutic targets [100] [101]. However, the clinical advancement of chaperone inhibitors has been significantly hampered by dose-limiting toxicities and a narrow therapeutic window [101]. These adverse effects are mechanistically linked to the fundamental biological roles of Hsp70, Hsp90, and related chaperones in normal cellular physiology. The constitutive expression of chaperones like Hsp90β and Hsp70 (Hsc70) is critical for vital signal transduction pathways in healthy cells; consequently, their broad inhibition disrupts essential processes, leading to on-target toxicity [100] [17]. This whitepaper delineates the mechanisms underlying these toxicities and synthesizes current strategies to optimize the therapeutic window of chaperone inhibition, with a specific focus on the integrated roles of the Hsp70 and Hsp90 systems in proteostasis.
Mechanisms of Toxicity in Chaperone Inhibition
On-Target Toxicities from Broach Proteostasis Disruption
The toxicity profile of first-generation Hsp90 inhibitors illustrates the consequences of broad proteostasis disruption. Geldanamycin and its analog 17-AAG induced significant hepatotoxicity, which was attributed to their non-selective inhibition of Hsp90 isoforms and subsequent degradation of essential client proteins in normal tissues [101]. A primary mechanistic driver of toxicity is the induction of the cytoprotective heat shock response (HSR). Upon inhibition of Hsp90's ATPase activity, the transcription factor HSF-1 is liberated and activated, leading to the compensatory upregulation of a suite of heat shock proteins, including Hsp70 and Hsp27 [102] [101]. This survival response blunts the antitumor efficacy of the inhibitors and can contribute to therapeutic resistance. The HSR is a conserved feedback mechanism wherein Hsp70 plays a key regulatory role; under normal conditions, Hsp70 binds to and inhibits HSF-1, but when Hsp70 is engaged by misfolded proteins resulting from Hsp90 inhibition, HSF-1 is activated [103] [17].
Isoform Non-Selectivity and Associated Toxicities
The human Hsp90 family consists of four major isoforms: the cytosolic Hsp90α (inducible) and Hsp90β (constitutive), the endoplasmic reticulum-localized GRP94, and the mitochondrial TRAP1 [100] [104]. Early, non-selective inhibitors target a highly conserved ATP-binding pocket across all isoforms, leading to widespread client protein degradation and mechanistic toxicity. For instance, the second-generation inhibitor luminespib (AUY922) caused visual disturbances and other side effects that limited its long-term administration, while ganetespib was associated with gastrointestinal toxicities and elevated liver enzymes [101]. These findings underscore that inhibiting the constitutive Hsp90β isoform, which is essential for fundamental cellular processes in non-malignant cells, is a major contributor to the narrow therapeutic window observed in clinical trials.
Strategic Optimization of the Therapeutic Window
Targeting Protein-Protein Interactions (PPIs) for Selective Disruption
A paradigm-shifting strategy to circumvent the limitations of ATP-competitive inhibitors involves targeting specific protein-protein interactions within the chaperone network. The interaction between Hsp90 and its kinase-specific co-chaperone Cdc37 is a particularly promising target [102]. Cdc37 acts as a scaffold that recruits and stabilizes a large subset of protein kinase clients for Hsp90-mediated maturation. Disrupting the Hsp90-Cdc37 interface offers several advantages for therapeutic window optimization, as detailed in Table 1.
Table 1: Key PPI-Targeted Strategies for Managing Chaperone Inhibition Toxicity
| Strategy | Molecular Target | Mechanistic Rationale | Representative Agents | Impact on Therapeutic Window |
|---|---|---|---|---|
| Co-chaperone PPI Disruption | Hsp90-Cdc37 interface [102] | Selectively destabilizes kinase clients (e.g., Akt, Raf) without globally impairing Hsp90 function. | Celastrol, Withaferin A, synthetic small molecules [102] | Minimizes HSR induction; reduces degradation of non-kinase clients in healthy tissues. |
| Isoform-Selective Inhibition | Unique structural pockets of GRP94 or TRAP1 [104] [101] | Exploits differential expression and clientele of organelle-specific Hsp90 isoforms. | MitoQ (TRAP1-targeting) [104] | Confines inhibition to tumor-specific proteostatic pathways (e.g., mitochondrial metabolism). |
| PROTAC-Based Degradation | E3 Ligase Recruitment to Hsp90 [101] | Induces proteasomal degradation of Hsp90 itself or specific oncogenic client proteins. | HSP90-PROTAC molecules [101] | Offers catalytic mode of action and potential for enhanced selectivity through ternary complex formation. |
The core logic is that PPI disruptors do not block the global ATPase activity of Hsp90 but rather interfere with a specific functional module. This selectivity means that the maturation of many non-kinase client proteins, which may be critical for normal cellular function, can proceed via alternative co-chaperones, thereby preserving vital proteostatic functions in healthy cells [102]. Preclinical studies confirm that Hsp90-Cdc37 disruptors effectively degrade oncogenic kinases and induce apoptosis in cancer cells without triggering a robust heat shock response, a key differentiator from N-terminal inhibitors [102].
Combination Therapies to Mitigate Toxicity and Overcome Resistance
Combining lower, less toxic doses of Hsp90 inhibitors with other anticancer agents presents a clinically viable path to enhance efficacy while managing the therapeutic window. The synergistic mechanisms of such combinations are multifaceted, as illustrated below.
Diagram 1: Synergistic mechanisms of HSP90 inhibitor-based combination therapies. Combining HSP90 inhibitors with other agents simultaneously targets multiple survival pathways, reducing the required dose and associated toxicity of the HSP90 inhibitor itself.
Rational combination partners include:
- Targeted Therapies: Co-administration with kinase inhibitors (e.g., EGFR, ALK inhibitors) prevents the activation of bypass signaling pathways that can cause resistance to monotherapy [101].
- Immunotherapies: Combination with immune checkpoint inhibitors (e.g., anti-PD-1/PD-L1) can reverse the immunosuppressive tumor microenvironment and enhance T-cell-mediated tumor cell killing [101].
- Chemotherapies: Synergy with agents like taxanes or gemcitabine enhances DNA damage and cell cycle arrest, allowing for dose reduction of both drugs [101].
This approach directly addresses the clinical challenge of dose-limiting toxicity by enabling the use of lower, better-tolerated doses of the Hsp90 inhibitor while achieving superior antitumor activity through synergistic mechanisms.
Experimental Toolkit for Evaluating Toxicity and Efficacy
Core Assays and Workflows
A critical component of optimizing the therapeutic window is the rigorous preclinical assessment of toxicity and efficacy. The following workflow outlines a standard protocol for evaluating novel Hsp90 inhibitors, from in vitro screening to in vivo validation.
Diagram 2: A sequential workflow for the preclinical evaluation of HSP90 inhibitors. This pipeline integrates in vitro and in vivo assessments to comprehensively profile efficacy and potential toxicity.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their applications in experimental protocols for investigating chaperone inhibition.
Table 2: Research Reagent Solutions for Chaperone Inhibition Studies
| Research Reagent / Tool | Function and Application in Experimental Protocols |
|---|---|
| Recombinant Hsp90 Isoforms (Hsp90α, Hsp90β, GRP94, TRAP1) | Used in surface plasmon resonance (SPR) and biochemical ATPase assays to determine binding affinity and inhibitory potency of novel compounds against specific isoforms [104]. |
| Cdc37 (N-terminal domain) Protein | Essential for co-crystallization studies and developing ELISA-based competitive binding assays to identify and characterize Hsp90-Cdc37 PPI disruptors [102]. |
| HSF-1 Activation Reporter Cell Line | A genetically engineered cell line (e.g., expressing luciferase under an HSE promoter) used to quantify heat shock response induction, a key indicator of potential toxicity [101]. |
| Isoform-Selective Chemical Probes (e.g., MitoQ for TRAP1) | Tool compounds used to dissect the specific biological functions of individual Hsp90 isoforms and validate isoform-specific targeting strategies [104]. |
| HSP70/HSP27 ELISA Kits | Validate target engagement and monitor the heat shock response by quantifying the upregulation of these proteins in cell lysates or patient serum following Hsp90 inhibition [103] [101]. |
| Client Protein Antibodies (e.g., against Her2, Akt, Raf) | Used in Western blot and immunofluorescence protocols to demonstrate on-target mechanism of action via degradation of specific Hsp90 client proteins in treated cells [100] [102]. |
Detailed Protocol: Hsp90-Cdc37 PPI Disruption Assay
- Incubation: Combine 1 µM of recombinant Hsp90 N-terminal/Middle domain with 1 µM of Cdc37 N-terminal domain in assay buffer (20 mM HEPES, pH 7.5, 50 mM KCl, 5 mM MgCl₂) in the presence of the test compound (e.g., 10 µM celastrol) or DMSO control. Incubate at 30°C for 60 minutes [102].
- Cross-linking: Add a chemical cross-linker (e.g., BS³ at a final concentration of 1 mM) to stabilize protein complexes for analysis. Quench the reaction after 30 minutes.
- Analysis: Resolve the cross-linked products via SDS-PAGE and perform Western blotting. Probe with anti-Hsp90 and anti-Cdc37 antibodies. A reduction in the intensity of the Hsp90-Cdc37 complex band in the treated sample compared to the DMSO control indicates successful disruption of the PPI [102].
Optimizing the therapeutic window of chaperone inhibition demands a shift from broad, ATP-competitive targeting to sophisticated, selective modulation. Strategies focused on disrupting specific PPIs, such as the Hsp90-Cdc37 complex, and employing rational combination therapies hold significant promise for mitigating the on-target toxicities that have limited earlier drug candidates. The future of this field lies in leveraging structural biology insights to design next-generation molecules, such as isoform-specific inhibitors and PROTACs, and in the clinical identification of robust biomarkers to select patient populations most likely to benefit from these targeted interventions. As our understanding of the integrated Hsp70 and Hsp90 chaperone networks in proteostasis deepens, so too will our ability to precisely inhibit their oncogenic functions while sparing their essential roles in normal physiology, ultimately achieving a wider, more effective therapeutic window.
Biomarker Development for Patient Stratification and Treatment Response Monitoring
Within the complex machinery of cellular proteostasis, molecular chaperones play an indispensable role in maintaining protein homeostasis by facilitating the folding, assembly, translocation, and degradation of proteins. Heat shock proteins Hsp70 and Hsp60 are central components of this chaperone network, with their expression and function frequently dysregulated in pathological states including cancer, neurodegenerative disorders, and cardiovascular diseases [17]. This technical guide examines the strategic development of Hsp70 and Hsp60 as clinical biomarkers, focusing on their application in patient stratification and longitudinal monitoring of therapeutic response. The dual localization of these chaperones—operating intracellularly to maintain proteostasis and appearing extracellularly as signaling molecules—creates unique opportunities for multi-compartment biomarker development. We present structured experimental data, methodological protocols, and analytical frameworks to advance the translation of chaperone biomarkers into clinical tools.
Hsp70 and Hsp60 in Cellular Proteostasis
Structural and Functional Organization of Hsp70
The Hsp70 family comprises highly conserved molecular chaperones ubiquitously expressed in prokaryotic and eukaryotic organisms. In humans, approximately 17 genes encode multiple Hsp70 proteins, including stress-inducible Hsp70s and constitutively expressed Hsp70 cognates (Hsc70) [17]. These isoforms distribute throughout cellular compartments—cytosol, nucleus, endoplasmic reticulum, and mitochondria—maintaining dynamic balance in protein synthesis, folding, degradation, and translocation.
Hsp70 proteins share conserved structural features critical to their chaperone function. The N-terminal nucleotide-binding domain (NBD, ~45 kDa) contains an ATPase activity that provides essential energy for conformational changes. The C-terminal substrate-binding domain (SBD, ~25 kDa) consists of a β-sandwich subdomain (SBDβ) that recognizes hydrophobic segments of client proteins and an α-helical "lid" (SBDα) that regulates substrate binding and release. These domains connect through a flexible linker and terminate in a conserved EEVD motif (Glu-Glu-Val-Asp) that mediates interactions with various co-chaperones [19] [17]. The allosteric regulation between NBD and SBD, driven by ATP hydrolysis, enables Hsp70 to cycle between low-affinity (ATP-bound) and high-affinity (ADP-bound) states for substrates, facilitating client protein folding, refolding, and complex assembly.
Hsp60 Chaperonin System
The Hsp60 chaperonin family, exemplified by the bacterial GroEL-GroES system, forms large double-ring complexes that provide an isolated compartment for protein folding. These ATP-dependent chaperonins function as essential "foldases," particularly for proteins difficult to fold in the crowded cellular environment. The human mitochondrial Hsp60-Hsp10 system shares this fundamental mechanism, assisting in the folding of imported proteins and maintaining mitochondrial proteostasis. Dysfunction in Hsp60 networks correlates with protein aggregation diseases, though its biomarker applications remain less explored than Hsp70.
Hsp70 as a Versatile Clinical Biomarker: Quantitative Evidence
Membrane Hsp70 (mHsp70) as a Tumor-Specific Marker
The selective presentation of Hsp70 on the plasma membrane of malignant cells—but not normal cells—provides a foundation for its use as a tumor-specific biomarker. Unlike intracellular Hsp70, which is present in most nucleated cells under stress, membrane Hsp70 (mHsp70) appears exclusively on tumor cells across diverse cancer types.
Table 1: mHsp70 Expression in Tumor versus Normal Tissues
| Tissue Type | Patient Population | mHsp70 Positive Cells (Mean %) | Assessment Method | Citation |
|---|---|---|---|---|
| Squamous cell carcinoma of head and neck (SCCHN) | 21 patients | 38% | Flow cytometry with cmHsp70.1 mAb | [105] |
| Normal connective tissue | 7 donors | 13% | Flow cytometry with cmHsp70.1 mAb | [105] |
This tumor-specific membrane localization enables mHsp70 detection for cancer diagnosis and circulating tumor cell (CTC) isolation. Comparative studies demonstrate that mHsp70-based CTC isolation outperforms EpCAM-based approaches, particularly following epithelial-to-mesenchymal transition (EMT) where EpCAM expression is often downregulated while mHsp70 remains stable [106].
Circulating Hsp70 as a Predictive and Monitoring Biomarker
Viable mHsp70-positive tumor cells actively release Hsp70 in lipid microvesicles, providing a measurable circulating biomarker. The compHsp70 ELISA detects this vesicular Hsp70 in patient blood, with levels correlating with tumor burden and therapeutic response.
Table 2: Circulating Hsp70 as a Predictive Biomarker in Cancer Patients
| Cancer Type | Patient Cohort | Hsp70 Measurement | Key Findings | Clinical Application | Citation |
|---|---|---|---|---|---|
| Non-metastatic breast cancer | 35 patients | compHsp70 ELISA during/following therapy | Progressive increase predicted therapeutic failure 2 years post-diagnosis | Early prediction of treatment resistance | [106] |
| Squamous cell carcinoma of head and neck (SCCHN) | 21 patients | Serum Hsp70 ELISA before/after radiotherapy | Significant drop in Hsp70 post-therapy in relapse-free patients; association with tumor volume | Therapy response monitoring | [105] |
| Various advanced cancers (prostate, lung, H&N) | 62 patients | compHsp70 ELISA | Correlation with gross tumor volume in NSCLC | Tumor burden assessment | [106] |
Notably, in atrial fibrillation, HSP27 (a small HSP) showed potential as a predictive biomarker despite no baseline association with AF stage or recurrence. HSP27 levels significantly increased in follow-up samples of patients with AF recurrence after pulmonary vein isolation, suggesting utility in predicting arrhythmia recurrence after ablative therapy [107].
Experimental Methodologies for Chaperone Biomarker Development
mHsp70 Detection on Tumor Cells
Protocol: Flow Cytometric Analysis of mHsp70 Expression
- Sample Preparation: Obtain fresh tumor biopsies (few mm³) and reference tissues during surgical resection. Prepare single-cell suspensions by mechanical disintegration through sterile mesh (75μm).
- Cell Staining: Wash 1×10⁵ cells with PBS containing 10% FCS. Incubate with FITC-conjugated cmHsp70.1 monoclonal antibody (or isotype-matched IgG1 control) on ice for 30 minutes in the dark.
- Analysis: Add propidium iodide to exclude dead cells. Analyze viable cells using flow cytometry (e.g., FACSCalibur). Calculate mHsp70-positive percentage by subtracting isotype control staining [105].
Technical Considerations: The cmHsp70.1 monoclonal antibody specifically recognizes the membrane-bound form of Hsp70. Immediate processing of fresh tissues maintains epitope integrity. Include both positive (known mHsp70+ cell line) and negative (normal tissue) controls in each experiment.
Circulating Hsp70 Measurement
Protocol: compHsp70 ELISA for Vesicular Hsp70
- Plate Coating: Incubate 96-well MaxiSorp plates with cmHsp70.2 coating antibody (1μg/mL in 0.1M carbonate/bicarbonate buffer, pH 9.6) overnight at room temperature.
- Blocking: Wash plates with PBS containing 0.05% Tween-20, then block with liquid plate sealer for 30 minutes to prevent non-specific binding.
- Sample Incubation: Add patient plasma samples (diluted 1:5 in StabilZyme Select Stabilizer) and incubate.
- Detection: Follow standard ELISA detection steps with appropriate secondary antibodies and substrates. Include recombinant Hsp70 for standard curve generation [106].
Technical Considerations: This ELISA specifically detects vesicular Hsp70 actively released by viable tumor cells, distinguishing it from free Hsp70 released during necrotic cell death. Sample integrity requires careful blood processing: centrifuge EDTA blood at 1500×g for 15 minutes at room temperature, then immediately freeze plasma at -80°C in aliquots.
Hsp70-Based Circulating Tumor Cell Isolation
Protocol: CTC Enrichment Using mHsp70 Antibodies
- Sample Collection: Draw peripheral blood (2×7.5mL) into EDTA tubes from patients with metastatic cancer.
- Immunomagnetic Separation: Incubate blood with magnetic beads conjugated with cmHsp70.1 monoclonal antibody.
- CTC Enumeration: Isplicate bead-bound cells and characterize using immunohistochemistry for cytokeratin (epithelial marker) and CD45 (leukocyte marker). Cytokeratin-positive/CD45-negative cells are classified as CTCs [106].
Technical Considerations: mHsp70-based CTC isolation demonstrates superiority over EpCAM-based approaches, particularly in cancers with mesenchymal features or following EMT where EpCAM expression decreases while mHsp70 remains stable.
Hsp70 in Immunological Regulation and Therapeutic Implications
The biomarker utility of Hsp70 extends beyond simple quantification to functional assessment of immune responses. Extracellular Hsp70 (eHsp70) exhibits dual immunoregulatory functions—acting as a damage-associated molecular pattern (DAMP) that activates innate immunity while simultaneously capable of suppressing excessive inflammation.
Diagram 1: Dual Immunoregulatory Functions of Extracellular HSP70. eHSP70 activates immune responses through TLR2/4 signaling and immunogenic cell death, while simultaneously suppressing immunity via Treg differentiation. Created with DOT language.
This immunological duality creates a complex biomarker landscape where Hsp70 levels must be interpreted within specific pathological contexts. In cancer, the balance between immunostimulatory and immunosuppressive Hsp70 functions may predict response to immunotherapy and guide patient stratification.
Research Reagent Solutions for Chaperone Biomarker Studies
Table 3: Essential Research Reagents for Hsp70 Biomarker Development
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Hsp70 monoclonal antibodies | cmHsp70.1 (FITC-conjugated) | Flow cytometry detection of mHsp70 on tumor cells | Recognizes membrane-bound form; used for CTC isolation |
| Hsp70 monoclonal antibodies | cmHsp70.2 | Coating antibody for compHsp70 ELISA | Specific for vesicular Hsp70 detection |
| ELISA kits | Duoset Hsp70 Immunoassay (R&D Systems, DYC1663) | Detection of soluble Hsp70 in serum/plasma | Modified protocol for serum samples; incubation at 4°C overnight |
| Cell isolation systems | Ficoll-Paque density gradient | Peripheral blood mononuclear cell (PBMC) isolation | Basis for CTC enrichment protocols |
| Magnetic bead systems | EpCAM mAb-coated beads | Comparative CTC isolation | Lower efficiency than mHsp70 approach post-EMT |
| Magnetic bead systems | cmHsp70.1 mAb-coated beads | High-efficiency CTC isolation | Superior for mesenchymal CTCs |
| Sample collection tubes | EDTA S-Monovettes (Sarstedt) | Blood collection for Hsp70 analysis | Maintains sample integrity for vesicular Hsp70 |
| Protein standards | Recombinant Hsp70 (Enzo Life Sciences, ADI-NSP-555) | ELISA standard curve; antibody detection | Quality control for assay performance |
The integration of Hsp70 and Hsp60 biomarkers into clinical development pipelines offers significant potential for advancing precision medicine. The consistent overexpression of mHsp70 across tumor types, the detectability of circulating vesicular Hsp70, and the functional role of extracellular Hsp70 in immune regulation create a multi-dimensional biomarker platform. Future development should focus on standardizing detection assays across platforms, establishing disease-specific cutoff values, and validating predictive algorithms that incorporate chaperone biomarkers with conventional clinical parameters. The dynamic nature of chaperone expression in response to therapeutic intervention positions these biomarkers as valuable tools for adaptive therapy strategies that evolve with changing tumor biology. As research continues to elucidate the complex chaperone networks in proteostasis, the biomarker applications of Hsp70 and Hsp60 will expand, ultimately enhancing patient stratification and enabling more precise monitoring of treatment responses across diverse disease states.
Functional Specialization, Disease Validation, and Network Integration
Within the cellular proteostasis network, the Hsp70 and Hsp60 chaperone families represent two fundamental, yet mechanistically distinct, pillars of protein folding and quality control. Hsp70 functions as a central triage hub, making ATP-dependent decisions on the folding, trafficking, or degradation of a diverse clientele through transient interactions. In contrast, Hsp60 operates as an enclosed folding chamber, providing a protected environment for the ATP-driven folding of proteins within its central cavity. This whitepaper delineates the structural and functional divergence between these systems, underscoring their unique yet complementary roles in proteostasis. Understanding these distinct mechanisms provides a critical framework for developing targeted therapeutic strategies for cancer, neurodegenerative disorders, and other diseases of proteostasis imbalance.
Protein homeostasis, or proteostasis, is essential for cellular viability and function, relying on a network of molecular chaperones to ensure the correct folding, localization, and turnover of proteins [108]. The Hsp70 and Hsp60 (chaperonin) families are among the most conserved and functionally critical components of this network. Despite sharing an overall goal of preventing protein misfolding and aggregation, they have evolved divergent structural mechanisms to manage client proteins. Hsp70 acts as a versatile triage factor, interacting with short, extended hydrophobic segments of clients to make folding decisions, often in collaboration with other machineries like Hsp90 and the proteasome [109] [108]. Conversely, Hsp60 functions as a specialized folding chamber, encapsulating entire proteins within an isolated barrel-shaped complex to facilitate folding in an ATP-dependent manner [1] [27]. This guide provides an in-depth technical comparison of their mechanisms, supported by experimental data and methodologies relevant to current proteostasis research.
Core Mechanisms: A Comparative Analysis
Hsp70: The ATP-Dependent Triage Hub
The Hsp70 chaperone system functions as a central decision-making point in the proteostasis network, directing client proteins to various fates including de novo folding, membrane targeting, or degradation [108].
- Allosteric Mechanism and ATPase Cycle: Hsp70's function is governed by an allosteric ATPase cycle. The chaperone consists of a Nucleotide-Binding Domain (NBD) and a Substrate-Binding Domain (SBD) [110]. In the ATP-bound state, the SBD has a low affinity for substrates, allowing rapid binding and release. ATP hydrolysis, stimulated by interaction with Hsp40 co-chaperones (J-domain proteins) and the client protein itself, triggers a conformational change. This transition to the ADP-bound state dramatically increases Hsp70's affinity for the client, stabilizing it and preventing aggregation [110] [108]. Subsequent nucleotide exchange, facilitated by Nucleotide Exchange Factors (NEFs) like Bag proteins or HspBP1, returns Hsp70 to the ATP-state, releasing the folded client or handing it off to another system [110].
- Triage Decisions: The fate of an Hsp70-bound client is determined by the cohort of co-chaperones with which Hsp70 interacts. For instance, interaction with the TPR-domain co-chaperone CHIP (an E3 ubiquitin ligase) directs clients towards proteasomal degradation. In contrast, collaboration with Hsp90 via the HOP/Sti1 co-chaperone facilitates the maturation of specific signaling proteins like kinases and steroid hormone receptors [109] [108]. This makes Hsp70 a critical node in cellular quality control.
Hsp60/Chaperonin: The Folding Nano-Cage
The Hsp60 family, or chaperonins, provide a physically segregated environment for protein folding, isolating clients from the crowded cytosol to prevent aggregation.
- Group I Chaperonin Structure: Mitochondrial Hsp60 (HSPD1) is a Group I chaperonin, structurally homologous to the bacterial GroEL. It typically forms a large complex comprising two stacked heptameric rings, creating a double-ring barrel with a central cavity in each ring [1] [27]. Each subunit is composed of three domains: apical (binds substrate and co-chaperone), intermediate (a hinge), and equatorial (contains ATPase activity) [27].
- Folding Cycle with a Lid: The folding reaction requires ATP and a dedicated co-chaperone, Hsp10 (GroES in bacteria), which acts as a "lid". The cycle begins with an unfolded protein binding to the hydrophobic apical domains of one ring (the cis ring) of Hsp60. Binding of Hsp10 and ATP to the same cis ring induces a massive conformational change: the apical domains twist and elevate, and the hydrophobic substrate-binding sites are rotated inward and replaced with hydrophilic residues. This encapsulates the substrate in an isolated folding chamber [1] [27]. After a folding period (seconds to minutes), ATP hydrolysis and binding of a new substrate to the opposite (trans) ring triggers the release of Hsp10, ADP, and the folded protein. If folding fails, the protein can be rebound for another attempt [27].
Table 1: Key Functional Parameters of Hsp70 and Hsp60 Systems
| Parameter | Hsp70 System | Hsp60 (Group I) System |
|---|---|---|
| Primary Function | Triage: folding, trafficking, degradation decision point | Compartmentalized folding of single proteins |
| Core Components | Hsp70, Hsp40 (JDP), NEF (e.g., Bag, HspBP1) | Hsp60 (Cpn60) double-ring, Hsp10 (Cpn10) lid |
| ATP Dependency | Yes; regulates substrate binding affinity | Yes; drives large conformational changes & encapsulation |
| Co-chaperone Role | Hsp40 stimulates ATPase; NEF promotes nucleotide release | Hsp10 forms lid structure for encapsulation |
| Typical Client Size | Short, extended hydrophobic peptides (7-10 aa) | Full proteins (15-60 kDa) [27] |
| Structural Outcome | Local unfolding, prevents aggregation, hands off clients | Global folding in an isolated environment |
Experimental Methodologies for Chaperone Analysis
Analyzing Hsp70-Hsp60 Protein Interactions
Direct molecular interactions between chaperones, such as mortalin (mtHsp70) and Hsp60, can be characterized using the following protocol adapted from co-immunoprecipitation studies [111].
Protocol: Co-immunoprecipitation of Mortalin and HSP60
- Cell Lysis: Harvest cultured cells (e.g., COS7) and lyse using NP-40 lysis buffer.
- Antibody Incubation: Incubate 400 µg of total cell lysate protein with control IgG, anti-HSP60 antibody, or anti-mortalin antibody overnight at 4°C with gentle agitation.
- Complex Pull-Down: Add Protein A–agarose beads (20 µL) and incubate for 30 minutes at 4°C to capture the antibody-protein complexes.
- Washing: Pellet beads by centrifugation (10,000 rpm, 2 minutes, 4°C) and wash thoroughly with NP-40 lysis buffer to remove non-specifically bound proteins.
- Western Blot Analysis: Elute bound proteins from beads and separate by SDS-PAGE. Transfer to a membrane and probe with antibodies against the putative binding partner (e.g., anti-mortalin if the IP was for HSP60, and vice versa) to confirm interaction [111].
Domain Mapping with Deletion Mutants: To identify interaction domains, recombinant GST-tagged mortalin deletion mutants can be generated. These purified proteins are incubated with cell lysate, followed by immunoprecipitation with an anti-HSP60 antibody. Western blotting with an anti-GST antibody reveals which mortalin fragments retain the ability to bind HSP60, pinpointing the critical interaction domain (e.g., the N-terminal region of mortalin) [111].
Functional Assays for Chaperone Activity
ATPase Activity Assay: The core functionality of Hsp70 is tied to its ATP hydrolysis rate. This can be measured using a coupled enzymatic assay or by detecting the release of inorganic phosphate (Pi) over time. The assay is performed with purified Hsp70, client protein, and its co-chaperone Hsp40. A key experiment is to demonstrate that Hsp40 and the client protein synergistically stimulate Hsp70's ATPase activity [110].
Client Refolding Assay: The functionality of the Hsp60 system is demonstrated by its ability to refold a denatured model substrate. A common substrate is chemically denatured rhodanese. 1. Denaturation: Incubate rhodanese in a denaturing buffer containing urea or guanidine HCl. 2. Initiate Refolding: Dilute the denatured rhodanese into a refolding buffer containing purified Hsp60, Hsp10, and ATP. 3. Measure Activity: After a set time, assay for recovered rhodanese enzymatic activity. Successful refolding is ATP-dependent and requires both Hsp60 and its co-chaperone Hsp10 [27].
Essential Research Reagents and Tools
Table 2: The Scientist's Toolkit: Key Reagents for Chaperone Research
| Reagent / Tool | Function / Application | Example & Notes |
|---|---|---|
| shRNA/siRNA Plasmids | Knockdown of chaperone expression to study functional loss. | Inducible shRNA vectors targeting HSP60; suppression leads to cancer cell growth arrest [111]. |
| Recombinant Proteins | In vitro binding and functional assays (ATPase, refolding). | GST-tagged mortalin deletion mutants for mapping protein interactions [111]. |
| Specific Antibodies | Detection, localization (immunofluorescence), and immunoprecipitation. | Anti-mortalin (e.g., monoclonal from Affinity Bioreagents) and anti-HSP60 (e.g., N-20, Santa Cruz) [111]. |
| Chemical Inhibitors | Probing chaperone function and therapeutic potential. | Small molecules targeting Hsp90 (e.g., 17-AAG), Hsp70, and Hsp60 are under investigation [6]. |
| Model Substrates | Assessing chaperone-mediated refolding activity. | Denatured rhodanese for Hsp60/GroEL refolding assays [27]. |
Integrated Chaperone Pathways in the Cell
The Hsp70 and Hsp60 systems, while functionally distinct, are integrated within the broader proteostasis network. Hsp70 often acts as a primary sensor for newly synthesized or stress-denatured proteins. Based on the co-chaperones it recruits, it can then direct clients to different fates. Hsp70's handoff of a client to Hsp90 for final maturation is one well-established branch. Similarly, clients that require the secluded environment of the chaperonin can be directed to Hsp60 for folding.
The diagram below illustrates the integrated decision-making process of the cellular chaperone network, highlighting the central triaging role of Hsp70 and the specialized folding function of Hsp60.
Clinical Implications and Therapeutic Targeting
The critical roles of Hsp70 and Hsp60 in cell proliferation and survival make them attractive therapeutic targets, particularly in oncology and neurodegenerative diseases.
- Cancer Therapeutics: Many cancer cells exhibit elevated expression of chaperones like Hsp90, Hsp70, and Hsp60, which they rely on to stabilize oncogenic clients and buffer proteotoxic stress [6]. Suppression of mortalin or Hsp60 expression via shRNA has been shown to induce growth arrest in cancer cells, validating them as targets [111]. Current drug development strategies are moving beyond pan-inhibitors to more selective approaches, including:
- Isoform-selective inhibitors to target specific chaperone isoforms.
- Protein-protein interaction (PPI) inhibitors designed to disrupt the critical interfaces between chaperones and their co-factors (e.g., Hsp70-Hsp40 or Hsp90-CDC37) [6].
- Neurodegeneration: Neurodegenerative diseases like Alzheimer's and Parkinson's are characterized by the accumulation of misfolded protein aggregates. Enhancing the activity of chaperone networks, including Hsp70 and Hsp60, is a promising strategy to promote the clearance of these toxic species and maintain neuronal health [1] [6].
Hsp70 and Hsp60 represent two evolutionarily ancient solutions to the fundamental biological challenge of protein folding. Hsp70 operates as a dynamic, ATP-controlled triage hub that interacts with a vast array of clients to direct their folding, localization, or degradation. In contrast, Hsp60 provides a dedicated, isolated folding chamber that promotes the native folding of proteins through ATP-driven encapsulation. Their non-redundant, and often collaborative, functions are indispensable for proteostasis. The continuing elucidation of their detailed mechanisms and structures, including high-resolution complex structures [6], provides a robust foundation for the rational design of novel therapeutics aimed at modulating the proteostasis network in human disease.
Cellular life depends on the accurate synthesis, folding, and maintenance of proteins. To maintain protein homeostasis (proteostasis), cells invest in an extensive proteostasis network (PN)—an integrated system of molecular machineries that coordinate protein synthesis, folding, conformational maintenance, and degradation [112]. This network is particularly crucial for preventing the accumulation of potentially toxic protein aggregates, a hallmark of aging and neurodegenerative diseases such as Alzheimer's and Huntington's disease [112] [113]. The PN comprises approximately 2,000 components in mammalian cells, organized into three major functional modules: protein synthesis, folding and conformational maintenance by molecular chaperones, and protein degradation via the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) [112]. Molecular chaperones, especially those from the Hsp70 and Hsp60 families, serve as central coordinators throughout the protein life cycle, from coordinating the folding of nascent chains emerging from ribosomes to directing the disassembly of toxic aggregates [112] [114] [115]. This review examines the cooperative integration of these PN components, with particular emphasis on the mechanistic insights into Hsp70 and Hsp60 chaperone function in maintaining proteome integrity.
Architectural Foundations of the Proteostasis Network
Modular Organization of the Proteostasis Network
The PN operates through functionally specialized yet highly interconnected modules. The Human Proteostasis Network Annotation consortium has systematically categorized these components into nine main branches: mitochondrial proteostasis, ER proteostasis, cytonuclear proteostasis, nuclear proteostasis, cytosolic translation, proteostasis regulation, extracellular proteostasis, the autophagy lysosome pathway (ALP), and the ubiquitin proteasome system (UPS) [116]. This sophisticated organization enables cells to maintain proteome balance across diverse cellular compartments and in the face of various endogenous and exogenous stresses.
Table 1: Core Functional Modules of the Human Proteostasis Network
| Module | Key Components | Primary Functions |
|---|---|---|
| Protein Synthesis | Ribosomes, tRNA, initiation/elongation factors, RQC complex | Polypeptide chain synthesis, co-translational folding, recognition of stalled translation |
| Chaperone-Mediated Folding | Hsp70 system (Hsp70, JDPs, NEFs), Hsp60/chaperonins, small Hsps | Nascent chain folding, conformational maintenance, prevention of aggregation |
| Protein Degradation | Ubiquitin-proteasome system (UPS), Autophagy-lysosomal pathway (ALP) | Clearance of misfolded/aggregated proteins, proteome renewal |
| Regulatory Systems | HSF1, UPRER, UPRmt | Transcriptional and translational control of PN components during stress |
The ribosome serves as the entry point into the PN, not only as a translation machine but as an active participant in protein folding. Recent evidence indicates that the ribosome plays a crucial role in cotranslational protein folding and solubility maintenance [117]. The ribosomal exit tunnel, approximately 80-100 Å long and 10-35 Å wide, typically accommodates 30-40 nascent residues and influences early folding events [117]. Beyond the tunnel, the highly negatively charged outer surface of the ribosome provides a platform where nascent chains of single-domain proteins become compact and acquire secondary and tertiary structure [117].
Quantitative Dimensions of the Proteostatic Challenge
The scale of the proteostatic challenge faced by cells is immense. An average human cell expresses approximately 10,000–13,000 different protein species with copy numbers varying over several orders of magnitude, from a few molecules to tens of thousands [112]. These proteins are synthesized at an average rate of five to six amino acids per second, generating more than a billion protein molecules per human cell with an average size of 560 amino acids [112]. This process is inherently error-prone, with a misincorporation rate of approximately 1 in 10⁴ amino acids, resulting in roughly 1 in 20 protein molecules containing a sequence error that potentially causes misfolding or reduced stability [112]. The PN must constantly surveil this complex proteomic landscape to prevent dysfunction.
Chaperone Systems: Mechanisms and Specificity
The Hsp70 Chaperone Network
The 70-kDa heat shock protein (Hsp70) chaperones are central components of the PN, facilitating the folding, assembly, membrane translocation, and quality control of proteins [24]. Hsp70s achieve their diverse functions through what has been termed "selective promiscuity"—interacting with a wide range of substrate proteins while minimizing undesired interactions [24]. The human Hsp70 system achieves remarkable substrate specificity through the coordinated action of 50 J-domain proteins (JDPs) and nucleotide exchange factors (NEFs) that regulate Hsp70's functional cycle [24].
JDPs can be categorized based on their substrate targeting mechanisms: "recruiters" that target Hsp70s to specific subcellular sites where substrates reside, "specialists" that bind substrates directly using highly specific binding sites, and "generalists" that employ multiple, versatile binding sites for broader substrate recognition [24]. This functional diversification enables the Hsp70 system to participate in diverse processes including Fe-S cluster biogenesis (through HSC20), clathrin uncoating (via auxilin), and protein translocation across endoplasmic reticulum and mitochondrial membranes [24].
The Hsp70 functional cycle involves ATP-dependent binding and release of extended segments of substrate proteins enriched in hydrophobic amino acids [118]. JDPs initially recognize and bind substrate proteins, then recruit Hsp70 and stimulate its ATPase activity, leading to tight binding to the substrate [24]. NEFs subsequently promote ADP release and ATP rebinding, resulting in substrate release [24]. This cycle allows Hsp70 to prevent aggregation and promote productive folding.
Table 2: Classification and Functions of J-Domain Proteins (JDPs) in Hsp70 System
| JDP Class | Representative Members | Targeting Mechanism | Cellular Functions |
|---|---|---|---|
| Class A | Hdj1, Hdj2 | Generalist substrate binding | Broad substrate recognition, de novo folding |
| Class B | Hdj4, Hdj5, Hdj7 | Specialist substrate binding | Specific client processing, organellar transport |
| Recruiters | Auxilin, ERj1, Zuotin | Location-specific Hsp70 recruitment | Clathrin uncoating, ER translocation, ribosome association |
Hsp60 Chaperonins: Compartmentalized Folding
The Hsp60 chaperonins, also known as chaperonins, form large barrel-shaped complexes that provide an isolated folding environment for proteins within the central cavity [114] [115]. Unlike Hsp70s that primarily interact with extended hydrophobic segments, chaperonins assist in the folding of entire protein domains through ATP-dependent cycles of substrate encapsulation and release [115]. This mechanism is particularly important for the folding of proteins with complex topologies or slow-folding kinetics that are prone to aggregation in the crowded cellular environment.
The cooperative action between Hsp70 and Hsp60 systems represents a key feature of the PN's functional architecture. Substrates may be initially stabilized by Hsp70 during or shortly after synthesis before transfer to chaperonins for complete folding, demonstrating the functional handoff mechanisms that enhance the efficiency and capacity of the PN [115].
Nascent Chain Biogenesis: Ribosomal Coordination with Chaperones
Ribosome-Assisted Folding and Chaperone Recruitment
The initial stages of protein folding begin during translation, with the ribosome serving as a critical platform for coordinating early folding events and chaperone recruitment. Research on ribosome-bound nascent chains (RNCs) of foldable proteins has revealed that specific interactions occur between nascent chains and ribosomal proteins near the exit tunnel [117]. Studies using E. coli flavohemoglobin (Hmp) RNCs demonstrated that shorter nascent chains (apoHmp1-140) interact primarily with ribosomal protein L23 near the exit tunnel [117]. As chains elongate (apoHmp1-189), they establish a wider interaction network that includes additional ribosomal proteins and molecular chaperones such as the trigger factor (TF) [117].
These ribosome-nascent chain interactions exhibit several important characteristics. First, they appear to be unbiased with respect to nascent chain sequence, charge, and hydrophobicity, suggesting the ribosome provides generalized thermodynamic assistance regardless of protein physicochemical properties [117]. Second, the interactions are dynamic and competitive—increasing concentrations of TF chaperone can partially or completely displace ribosomal proteins from nascent chains, indicating a handoff mechanism from ribosomal surveillance to dedicated chaperone systems [117].
Experimental Approaches for Studying Nascent Chain Interactions
Key insights into ribosome-nascent chain interactions have been obtained through sophisticated experimental methodologies:
- Chemical Crosslinking: Zero-length crosslinkers like 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) enable identification of direct protein-protein interactions between nascent chains and ribosomal components [117].
- Time-Resolved Fluorescence Anisotropy: This technique provides information on the dynamics and conformational freedom of specific regions within nascent chains [117].
- Puromycin Sensitivity Assays: Treatment with puromycin verifies the ribosome-bound status of nascent chains by causing premature chain release [117].
- Low-pH SDS-PAGE: This method maintains the integrity of acid-labile bonds in peptidyl-tRNA complexes, allowing analysis of RNCs [117].
Diagram Title: Nascent Chain Interactions Evolve with Length
Quality Control: Ribosome-Associated Pathways
Ribosome-Associated Quality Control (RQC) Complex
Errors during translation that cause ribosome stalling pose a significant threat to proteostasis, producing incomplete nascent chains that must be rapidly recognized and removed. The Ribosome-associated Quality Control (RQC) complex serves as a critical surveillance system for resolving these stalled translation events [119]. Recent structural studies of the yeast RQC complex using cryo-EM have revealed the mechanistic details of how this system orchestrates nascent chain removal.
The core RQC machinery includes several key components: Rqc2 recognizes aberrant 60S subunits bearing peptidyl-tRNA and initiates RQC complex assembly; Ltn1 is an E3 ubiquitin ligase that ubiquitylates the nascent chain; Rqc1 bridges the 60S subunit with ubiquitin and Ltn1; and the Cdc48-Ufd1-Npl4 complex functions as a segregase that extracts ubiquitylated peptides from the 60S ribosome [119]. A crucial finding is that Ltn1 directly recruits Cdc48 through interactions between Cdc48's N-terminal domain and a helical array in Ltn1, establishing Ltn1 as a bona fide Cdc48 adaptor [119].
Mechanism of Stalled Nascent Chain Processing
The RQC pathway operates through a carefully orchestrated sequence of events:
- Recognition: Rqc2 detects stalled 60S subunits with peptidyl-tRNA and initiates RQC assembly.
- CAT-tail Addition: Rqc2 mediates non-templated elongation of nascent polypeptides by recruiting alanine- and threonine-charged tRNAs, resulting in carboxy-terminal alanine and threonine (CAT) tails.
- Ubiquitylation: Ltn1, enhanced by Rqc1, assembles K48-linked polyubiquitin chains on exposed lysines within the exit tunnel-proximal portion of the nascent chain.
- Extraction: The Cdc48-Ufd1-Npl4 complex recognizes polyubiquitin chains and uses ATP hydrolysis to mechanically extract the nascent chain from the ribosomal exit tunnel.
- Degradation: The extracted polypeptide is delivered to the proteasome for degradation.
Mutations that disrupt the interaction between Ltn1 and Cdc48 (e.g., Ltn1 R945A, Q1022A, R1023A) significantly impair substrate degradation, confirming the functional importance of this recruitment mechanism [119].
Diagram Title: RQC-Mediated Processing of Stalled Nascent Chains
Aggregate Disassembly: Chaperone Mechanisms in Protein Disaggregation
The Bi-Chaperone Disaggregase System
When prevention mechanisms fail and protein aggregates form, cells employ specialized disaggregation systems to dismantle these potentially toxic structures. The core of this disaggregation activity in many organisms centers on a bi-chaperone system involving Hsp70 and Hsp100 chaperones [118]. In E. coli, this system comprises the Hsp70 DnaK and the Hsp100 ClpB, which collaborate to recognize, disentangle, and refold aggregated proteins.
The current model for disaggregation proposes that DnaK first binds to exposed hydrophobic segments of aggregated proteins, followed by recruitment of ClpB oligomers [118]. Subsequent transfer of the substrate from DnaK to ClpB activates the ATP-dependent threading activity of ClpB, which processively translocates polypeptide chains through its central pore, effectively disentangling aggregated material [118]. This mechanism demonstrates the functional synergy between different chaperone classes in resolving complex proteostatic challenges.
Structural Determinants of Aggregate Disassembly
Surprisingly, the efficiency of chaperone-mediated disaggregation depends less on the overall size of aggregate particles and more on their internal structural properties [118]. Research using ribulose-1,5-carboxylase oxygenase (RuBisCO) as a model substrate revealed that structurally distinct aggregates formed from the same protein exhibit dramatically different susceptibilities to disaggregation, despite similar particle sizes and chaperone binding capacities [118].
Two aggregate types were characterized: fast-growing (F-type) aggregates enriched in β-sheet content with nascent amyloid character, and slow-growing (S-type) aggregates with different structural properties [118]. Importantly, these structural alterations progress with surprising speed, rendering aggregates resistant to disassembly within minutes of formation [118]. This highlights the critical importance of early intervention in preventing the accumulation of recalcitrant aggregates.
Experimental Analysis of Disaggregation Kinetics
Advanced biophysical techniques have been essential for elucidating the mechanisms of chaperone-mediated disaggregation:
- Burst Analysis Spectroscopy (BAS): A single particle fluorescence technique that enables population-resolved kinetics measurements of aggregate disassembly without ensemble averaging [118].
- Multi-Color BAS (MC-BAS): An extension that allows real-time monitoring of chaperone binding stoichiometries in distinct aggregate sub-populations [118].
- Thioflavin T (ThT) Fluorescence: Detects β-sheet content in aggregates, revealing structural differences between aggregate types [118].
- bis-ANS Fluorescence: Measures solvent-exposed hydrophobic surface area, providing information on aggregate surface properties [118].
Table 3: Characteristics of Structurally Distinct RuBisCO Aggregates
| Property | S-Type Aggregates | F-Type Aggregates | Measurement Technique |
|---|---|---|---|
| Formation Kinetics | Slow-growing | Fast-growing | Static light scattering |
| β-Sheet Content | Lower | Higher | Thioflavin T fluorescence |
| Surface Hydrophobicity | Moderate | Elevated | bis-ANS fluorescence |
| Disaggregation Efficiency | Higher | Lower | BAS with chaperones |
Integrated Crosstalk: Proteasome and Autophagy in Aggregate Clearance
The collaboration between proteostatic systems extends to the degradation machinery, where coordinated action between the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP) enables efficient clearance of protein aggregates. When aggregates cannot be processed by the proteasome directly, they are frequently concentrated into aggresomes, perinuclear inclusion bodies that are subsequently removed by autophagy [120].
Remarkably, proteasomes play a crucial activation role in aggresome clearance despite being unable to process aggregates directly. The proteasomal deubiquitinating enzyme Poh1 cleaves ubiquitinated proteins and releases unanchored K63-linked ubiquitin chains [120]. These free ubiquitin chains serve as signaling molecules that bind and activate the deacetylase HDAC6, which in turn stimulates actinomyosin- and autophagy-dependent aggresome processing [120]. This mechanism demonstrates how the PN employs metabolite-like signaling molecules to coordinate different proteostatic modules.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 4: Key Research Reagents for Proteostasis Network Studies
| Reagent/Technique | Key Function | Application Examples |
|---|---|---|
| Chemical Crosslinkers (EDC) | Zero-length crosslinking for protein interaction mapping | Identifying nascent chain-ribosomal protein interactions [117] |
| Thioflavin T (ThT) | Fluorescent dye reporting on β-sheet content | Characterizing amyloid-like structure in aggregates [118] |
| bis-ANS | Environment-sensitive fluorophore for hydrophobic surfaces | Probing exposed hydrophobic patches on aggregates [118] |
| Puromycin | Causes premature chain release from ribosomes | Verifying ribosome-bound status of nascent chains [117] |
| Burst Analysis Spectroscopy (BAS) | Single-particle fluorescence technique | Monitoring disaggregation kinetics without ensemble averaging [118] |
| Cryo-Electron Microscopy | High-resolution structural analysis | Determining architecture of RQC complexes [119] |
| ADP-BeFx | Non-hydrolyzable ATP analog | Trapping Cdc48 in active conformation for structural studies [119] |
The cooperative integration of proteostasis network components—from ribosomal coordination with nascent chains to chaperone-mediated disaggregation of aggregates—represents a fundamental biological principle for maintaining proteome health. The mechanistic insights into Hsp70 and Hsp60 function reveal how specificity emerges from promiscuous chaperone systems through sophisticated regulatory networks of co-chaperones and adaptor proteins. Understanding these integrated mechanisms provides crucial foundation for developing therapeutic interventions for age-related neurodegenerative diseases and other protein conformational disorders. Pharmacological approaches that enhance the capacity of specific PN modules, particularly those that boost disaggregation activity or stabilize the folding of metastable proteins, hold significant promise for delaying the onset of pathologies associated with proteostasis collapse [112] [113]. As structural and single-molecule techniques continue to advance, they will undoubtedly reveal further intricacies of how the PN maintains proteome balance throughout the cellular lifespan.
Molecular chaperones, particularly Hsp70 and Hsp60, play central yet paradoxical roles in cellular proteostasis. In cancer, these chaperones are co-opted to support tumor proliferation and survival, whereas in neurodegenerative proteinopathies, their dysregulation contributes to pathogenic protein aggregation. This whitepaper provides a technical analysis of the distinct mechanisms by which Hsp70 and Hsp60 operate in these contrasting disease contexts, supported by structured quantitative data, experimental methodologies, and visual signaling pathways. We further explore the therapeutic implications of targeting these chaperone systems, including current challenges and emerging strategies in drug development for oncology and neurology.
Molecular chaperones constitute a complex network of proteins dedicated to maintaining protein homeostasis (proteostasis) within the cell. As essential components of the protein quality control system, Hsp70 and Hsp60 chaperone families facilitate the folding of nascent polypeptides, prevent the aggregation of misfolded proteins, and assist in the degradation of damaged proteins [121] [6]. Under physiological stress conditions, including elevated temperature, oxidative stress, and metabolic alterations, the expression of these chaperones is significantly upregulated to ensure cellular survival [122] [71].
The critical importance of chaperone function becomes evident in disease states, where their roles diverge dramatically. In cancer biology, Hsp70 and Hsp60 are frequently overexpressed, promoting tumor cell proliferation, metastasis, and resistance to cell death [19] [122] [71]. Conversely, in neurodegenerative proteinopathies such as Alzheimer's disease (AD) and Parkinson's disease (PD), chaperone systems often become overwhelmed or dysfunctional, failing to prevent the accumulation of toxic protein aggregates that characterize these conditions [121] [58]. This whitepaper examines the dualistic nature of Hsp70 and Hsp60 chaperone mechanisms across these disease landscapes, providing a framework for targeted therapeutic interventions.
Hsp70 and Hsp60: Structural and Functional Mechanisms
Hsp70 Chaperone System
The Hsp70 family consists of highly conserved molecular chaperones present in various cellular compartments, including the cytosol, nucleus, endoplasmic reticulum, and mitochondria [19] [71]. In humans, 13 Hsp70 homologs are encoded by distinct genes located on multiple chromosomes [19].
Structural Organization: Hsp70 proteins share a conserved domain architecture comprising:
- An N-terminal Nucleotide-Binding Domain (NBD):
- Approximately 45 kDa in size
- Contains four subdomains (IA, IB, IIA, IIB) forming a cleft for ATP binding
- Hydrolyzes ATP to drive conformational changes
- A C-terminal Substrate-Binding Domain (SBD):
- ~25 kDa domain with β-sheet structure that binds client proteins
- Contains a helical lid subdomain (SBDα) that regulates substrate access
- A flexible linker region connecting NBD and SBD
- An EEVD motif at the extreme C-terminus that mediates interactions with co-chaperones [19] [71]
Functional Cycle: Hsp70 operates through an ATP-dependent cycle regulated by co-chaperones. In the ATP-bound state, Hsp70 exhibits low substrate affinity and fast binding/release kinetics. ATP hydrolysis, stimulated by J-domain proteins (Hsp40 family), transitions Hsp70 to the ADP-bound state with high substrate affinity and slow release kinetics. Nucleotide exchange factors (NEFs) then catalyze ADP-to-ATP exchange, resetting the cycle [19]. This coordinated mechanism allows Hsp70 to bind hydrophobic segments of client proteins, preventing misfolding and aggregation.
Hsp60 Chaperone System
Hsp60, also known as chaperonin, forms a large barrel-shaped complex that provides an isolated folding environment for client proteins.
Structural Organization: Hsp60 adopts a characteristic double-ring architecture with each ring comprising 7-8 subunits. Each monomer consists of three distinct domains:
- Equatorial Domain: Contains ATP binding sites and forms the ring interface
- Intermediate Domain: Forms a β-sheet structure creating a central chamber
- Apical Domain: Functions as a lid regulating substrate entry [71]
Functional Cycle: The Hsp60 chaperonin system functions in conjunction with its co-chaperone Hsp10. Without ATP, Hsp60 exists as a single-ring heptamer. ATP binding triggers double-ring formation, while ATP hydrolysis induces conformational changes that drive substrate folding and release [122] [71]. This complex provides an isolated environment conducive to proper protein folding, particularly for proteins vulnerable to aggregation.
Table 1: Structural and Functional Characteristics of Hsp70 and Hsp60
| Feature | Hsp70 | Hsp60 |
|---|---|---|
| Molecular Structure | Monomeric with NBD and SBD domains | Double-ring oligomeric complex |
| ATP Dependence | ATP-dependent | ATP-dependent |
| Cellular Localization | Cytosol, nucleus, ER, mitochondria | Mitochondria, cytosol |
| Co-chaperones | HSP40, Bag-1, Hip, Hop, CHIP | HSP10 |
| Primary Functions | Protein folding, refolding, translocation, degradation | Protein folding and assembly in isolated chamber |
| Client Spectrum | Unfolded polypeptides, transcription factors, kinases | Specific mitochondrial and cytosolic proteins |
Chaperone Functions in Cancer Proliferation
Oncogenic Mechanisms of Hsp70
In cancer systems, Hsp70 exhibits multifaceted pro-tumorigenic functions through several distinct mechanisms:
Anti-Apoptotic Signaling: Hsp70 directly interferes with both intrinsic and extrinsic apoptosis pathways. It binds to apoptosis protease activating factor-1 (Apaf-1), preventing the formation of the apoptosome complex and subsequent activation of caspase-9 [123]. Hsp70 also inhibits c-Jun N-terminal kinase (JNK) signaling and stabilizes lysosomes, providing comprehensive protection against programmed cell death [123].
Stabilization of Oncogenic Clients: Hsp70 interacts with and stabilizes numerous oncoproteins and signaling molecules that drive tumor progression. These client relationships enable cancer cells to maintain hyperactive growth signals and survival pathways despite genomic instability and proteotoxic stress [19] [71].
Immune Modulation: In the tumor microenvironment, extracellular Hsp70 activates immune cells including natural killer (NK) cells, suggesting potential for immunotherapeutic applications [19]. However, intracellular Hsp70 predominantly contributes to immune evasion by suppressing immunogenic cell death pathways.
Oncogenic Mechanisms of Hsp60
Hsp60 contributes to tumorigenesis through several established mechanisms:
Mitochondrial Anti-Apoptotic Regulation: Within mitochondria, Hsp60 interacts with and stabilizes proteins involved in apoptosis regulation, including direct inhibition of pro-apoptotic factors [122].
Pro-Inflammatory Signaling: Cytosolic Hsp60 directly interacts with IκB kinase (IKK), promoting activation of NF-κB-dependent gene transcription and establishing a pro-inflammatory microenvironment conducive to tumor growth [58].
Metabolic Reprogramming: Hsp60 supports the altered metabolic requirements of cancer cells by stabilizing mitochondrial enzymes involved in energy production and biosynthetic pathways.
Table 2: Hsp70 and Hsp60 in Cancer Hallmarks
| Cancer Hallmark | Hsp70 Mechanisms | Hsp60 Mechanisms |
|---|---|---|
| Sustained Proliferation | Stabilizes oncogenic clients (Raf, Akt) | Enhances mitochondrial metabolic enzymes |
| Evasion of Growth Suppression | Interacts with tumor suppressor p53 | Modulates cell cycle regulators |
| Resistance to Cell Death | Inhibits apoptosome formation; stabilizes lysosomes | Mitochondrial anti-apoptotic signaling |
| Activation of Invasion & Metastasis | Stabilizes matrix metalloproteinases | Promotes epithelial-mesenchymal transition |
| Tumor-Promoting Inflammation | Limited direct role | Activates NF-κB pro-inflammatory pathway |
| Deregulated Cellular Energetics | Limited direct role | Enhances mitochondrial metabolic efficiency |
Experimental Approaches in Cancer Chaperone Research
Client Protein Identification:
- Co-Immunoprecipitation (Co-IP): Incubate cell lysates with Hsp70/Hsp60 antibodies coupled to magnetic beads. Wash with mild buffer (25mM Tris, 150mM NaCl, 1% NP-40, pH 7.4). Elute bound complexes with low pH buffer (0.2M glycine, pH 2.5) and identify clients by mass spectrometry [124].
- Proximity Ligation Assay: Use species-specific primary antibodies against chaperones and putative client proteins with fluorescent-labeled secondary antibodies to visualize intracellular interactions [71].
Functional Validation:
- RNA Interference: Transfect cancer cells with siRNA targeting Hsp70/Hsp60 (50nM final concentration) using lipid-based transfection reagents. Assess impact on oncogenic signaling (72h post-transfection) and colony formation (14-day assay) [123].
- Inhibitor Studies: Treat 3D spheroid cultures with HSP90 inhibitors (17-AAG, 100nM) and measure compensatory Hsp70 induction by Western blot at 24h intervals [124] [123].
Chaperone Dysfunctions in Neurodegenerative Proteinopathies
Neuroprotective Mechanisms of Hsp70
In neuronal systems, Hsp70 provides crucial defense against protein misfolding through multiple mechanisms:
Direct Anti-Aggregation Activity: Hsp70 binds to aggregation-prone proteins including amyloid-β (AD), tau (AD), and α-synuclein (PD), preventing their transition into toxic oligomers and fibrils. This direct interaction shields exposed hydrophobic regions that would otherwise drive aberrant protein assembly [121] [5].
Protection Against Proteotoxic Stress: Under conditions of oxidative stress or metabolic impairment, Hsp70 expression is induced to refold damaged proteins and prevent widespread proteostasis collapse. This function is particularly critical in post-mitotic neurons, which cannot dilute accumulated damage through cell division [121].
Regulation of Protein Clearance: Hsp70 works cooperatively with protein degradation systems, including the ubiquitin-proteasome system and autophagy pathways, to target irreversibly damaged proteins for removal [121] [58].
Neuroprotective Mechanisms of Hsp60
Hsp60 contributes to neuronal proteostasis through mitochondrial-specific functions:
Mitochondrial Protein Quality Control: As a central component of mitochondrial proteostasis, Hsp60 facilitates the proper folding of nuclear-encoded proteins imported into mitochondria and assists in the assembly of mitochondrial enzyme complexes essential for energy production [122] [58].
Oxidative Stress Management: By maintaining the functional integrity of mitochondrial antioxidant enzymes, Hsp60 helps mitigate oxidative damage that contributes to neurodegenerative pathogenesis [58].
Chaperone Deficiencies in Neurodegeneration
Neurodegenerative conditions are characterized by specific failures in chaperone systems:
Alzheimer's Disease: Both Hsp70 and Hsp60 demonstrate impaired interactions with tau and amyloid-β, leading to increased aggregation propensity. Intracellular Hsp70 levels often become insufficient to cope with chronic proteotoxic stress, while extracellular Hsp70 and Hsp60 may acquire pro-inflammatory functions that exacerbate neuroinflammation [121] [58].
Parkinson's Disease: Hsp70 shows reduced binding affinity for misfolded α-synuclein, permitting its aggregation into Lewy bodies. The limited capacity of neuronal Hsp70 expression systems creates vulnerability to proteostatic challenge [121].
Amyotrophic Lateral Sclerosis: Mutant SOD1 aggregation exceeds Hsp70/Hsp60 refolding capacity, leading to motor neuron toxicity. Chaperone systems become progressively overwhelmed as disease advances [58].
Table 3: Hsp70 and Hsp60 Alterations in Neurodegenerative Proteinopathies
| Disease | Pathogenic Protein | Hsp70 Alterations | Hsp60 Alterations |
|---|---|---|---|
| Alzheimer's Disease | Aβ, tau | Insufficient clearance of Aβ and tau; extracellular pro-inflammatory role | Mitochondrial dysfunction; extracellular pro-inflammatory signaling |
| Parkinson's Disease | α-synuclein | Impaired binding to α-synuclein; failed prevention of Lewy body formation | Reduced mitochondrial protection; increased oxidative stress |
| Huntington's Disease | mutant huntingtin | Inadequate handling of polyQ-expanded protein; aggregation promotion | Compromised mitochondrial protein import and folding |
| Amyotrophic Lateral Sclerosis | mutant SOD1, TDP-43 | Overwhelmed by aggregation-prone clients; sequestration in inclusions | Loss of mitochondrial proteostasis; energy failure |
Experimental Approaches in Neurodegeneration Chaperone Research
Aggregation Inhibition Assays:
- Thioflavin T (ThT) Assay: Incubate 10μM amyloid-β1-42 or 15μM α-synuclein with varying Hsp70 concentrations (0.5-5μM) in aggregation buffer (50mM HEPES, 150mM NaCl, pH 7.4). Monitor ThT fluorescence (excitation 440nm/emission 485nm) every 5min for 24h with continuous shaking [121].
- Filter Trap Assay: Apply protein samples to cellulose acetate membrane under vacuum. Detect aggregated species using protein-specific antibodies after 1h blocking with 5% non-fat milk [121].
Functional Rescue Experiments:
- Viral Vector-Mediated Chaperone Expression: Deliver Hsp70 cDNA via AAV9 vector (titer ≥1×1012 vg/mL) to mouse hippocampus or striatum. Evaluate behavioral improvement (Morris water maze, rotarod) and pathological burden at 4-8 weeks post-injection [121].
- Chaperone Transduction: Fuse Hsp70 with cell-penetrating PEP-1 peptide at 1:10 molar ratio in PBS for 30min at 37°C. Apply to primary neuronal cultures (5μM final) and assess cytoprotection against proteotoxic insults [121].
Comparative Signaling Pathways: Visualizing Divergent Mechanisms
Hsp70 in Cancer vs. Neurodegeneration Signaling
Hsp60 in Cancer vs. Neurodegeneration Signaling
Therapeutic Targeting and Research Toolkit
Experimental Protocols for Chaperone Research
Hsp70 ATPase Activity Assay:
- Principle: Measure inorganic phosphate release during ATP hydrolysis
- Protocol:
- Prepare reaction mixture (25mM HEPES, 50mM KCl, 5mM MgCl₂, pH 7.4)
- Add 1μM Hsp70, 2μM Hsp40, and 2mM ATP
- Incubate at 37°C for 0, 15, 30, 45, 60min
- Stop reaction with 5% SDS
- Develop with malachite green reagent (0.03% malachite green, 1% ammonium molybdate, 0.5% Triton X-100)
- Measure A620nm and calculate phosphate using KH₂PO₄ standard curve [19]
Hsp60 Refolding Assay:
- Principle: Monitor reactivation of denatured enzyme with Hsp60/Hsp10 system
- Protocol:
- Denature 5μM mitochondrial malate dehydrogenase in 6M guanidine-HCl for 2h
- Rapidly dilute 1:100 into refolding buffer (50mM HEPES, 10mM MgCl₂, 10mM KCl, 2mM ATP, pH 7.4)
- Add 2μM Hsp60 and 4μM Hsp10
- Monitor enzyme activity at 340nm (NADH oxidation) every minute for 90min
- Compare initial velocities to native enzyme control [122]
Research Reagent Solutions
Table 4: Essential Research Reagents for Hsp70/Hsp60 Investigations
| Reagent/Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| HSP Inhibitors | 17-AAG, PU-H71, VER-155008 | Target validation, combination therapy studies | Inhibit ATPase activity; disrupt client-chaperone interactions |
| Expression Vectors | pcDNA3.1-Hsp70, pEGFP-Hsp60, AAV9-Hsp70 | Functional rescue experiments; overexpression studies | Enable chaperone expression in cellular and animal models |
| Antibodies | Anti-Hsp70 (clone C92F3A-5), Anti-Hsp60 (clone LK-1) | Western blot, immunohistochemistry, co-IP | Detect expression localization; isolate chaperone complexes |
| Cell Lines | SH-SY5Y (neurodegeneration), K562 (cancer), HEK293T (overexpression) | Disease modeling; mechanistic studies | Provide relevant cellular contexts for functional assays |
| Protein Aggregation Kits | Recombinant α-synuclein, Amyloid-β1-42, Thioflavin T | Aggregation inhibition assays | Enable quantification of chaperone anti-aggregation activity |
Therapeutic Development Strategies
Cancer-Targeted Approaches:
- Hsp90 Inhibition: Geldanamycin derivatives (17-AAG, 17-DMAG) indirectly target Hsp70 by inducing compensatory expression, creating proteostatic vulnerability in cancer cells [124] [123].
- Hsp70 Direct Inhibition: Small molecules like VER-155008 and PET-16 target Hsp70 ATPase activity, showing promise in preclinical models but facing challenges with therapeutic windows [124].
- Combination Therapies: Sequential administration of Hsp90 inhibitors followed by Hsp70 suppression prevents adaptive resistance mechanisms in tumor cells [123].
Neurodegeneration-Targeted Approaches:
- Hsp70 Induction: Hsp90 inhibitors such as 17-AAG indirectly boost Hsp70 levels, enhancing cellular capacity to handle proteotoxic stress [124] [58].
- Chaperone Gene Therapy: AAV-mediated Hsp70 delivery demonstrates neuroprotection in models of Parkinson's and Alzheimer's disease [121].
- Protein Transduction: Cell-penetrating peptides (PEP-1) fused to Hsp70 enable direct chaperone delivery across the blood-brain barrier [121].
The paradoxical roles of Hsp70 and Hsp60 in cancer proliferation versus neurodegenerative proteinopathies highlight the complex, context-dependent nature of chaperone biology in human disease. In cancer, these chaperones are harnessed to support survival and growth under proteotoxic stress, while in neurodegeneration, their diminished capacity or overwhelming burden enables pathogenic protein aggregation. Understanding these divergent mechanisms provides critical insights for developing targeted therapeutic strategies.
Future research directions should focus on:
- Isoform-Specific Targeting: Developing compounds that selectively target disease-associated chaperone isoforms or complexes
- Temporal Regulation: Precise control of chaperone induction timing to match therapeutic windows
- Combination Approaches: Rational polypharmacy that modulates multiple proteostasis network components simultaneously
- Biomarker Development: Identifying chaperone expression signatures that predict disease progression and treatment response
The continued elucidation of Hsp70 and Hsp60 mechanisms in proteostasis will undoubtedly yield novel therapeutic opportunities for addressing the fundamental protein folding defects that underlie both cancer and neurodegenerative disorders.
Heat shock protein 70 (Hsp70) exemplifies a paradigm of functional duality in immune regulation, operating as both a potent immune activator and suppressor through distinct molecular mechanisms and cellular contexts. This whitepaper examines Hsp70's complex roles within the broader framework of chaperone-mediated proteostasis, detailing its capacity to enhance antigen presentation and activate inflammatory pathways while simultaneously inducing immunosuppressive responses. Recent advances illuminate how Hsp70's intracellular and extracellular forms differentially influence immune outcomes through specific receptor interactions and signaling pathways. Understanding these mechanistic insights provides crucial foundations for developing Hsp70-targeted therapeutic strategies in cancer, autoimmune diseases, and immunomodulation, offering researchers and drug development professionals innovative approaches to harness this chaperone's dual nature for clinical applications.
Within the cellular chaperone network, Hsp70 represents a central node in maintaining proteostasis through its fundamental roles in protein folding, assembly, and degradation. The Hsp70 family consists of highly conserved molecular chaperones present across all organisms, with 13 members identified in humans that localize to various cellular compartments including the cytosol, endoplasmic reticulum, and mitochondria [19]. These proteins function as critical components of the protein quality control system, preventing misfolding and aggregation under both physiological and stress conditions [6]. The well-characterized inducible form, Hsp72, is strongly upregulated during cellular stress, while the constitutive Hsc70 is consistently expressed with moderate stress induction [125].
Hsp70's structural architecture enables its diverse functional capabilities, featuring an N-terminal nucleotide-binding domain (NBD) with ATPase activity and a C-terminal substrate-binding domain (SBD) connected by a flexible linker [126] [127]. This configuration allows Hsp70 to undergo ATP-dependent conformational changes that facilitate binding to client proteins, with substrate affinity regulated by co-chaperones including Hsp40 and nucleotide exchange factors (NEFs) [19]. Beyond these canonical proteostasis functions, Hsp70 has emerged as a significant immunomodulator, operating at the interface of innate and adaptive immunity through mechanisms that remain incompletely understood but offer promising therapeutic targets.
Structural Basis for Hsp70 Functionality
Domain Organization and Allosteric Regulation
The molecular function of Hsp70 relies on sophisticated allosteric regulation between its N-terminal and C-terminal domains, driven by ATP-ADP conversion [126] [128]. The 45 kDa N-terminal nucleotide-binding domain (NBD) consists of four subdomains (IA, IB, IIA, IIB) that form a binding cleft for ATP coordination, requiring Mg²⁺ and K⁺ ions for stabilization [19]. The 25 kDa C-terminal substrate-binding domain (SBD) contains a β-sandwich structure with a hydrophobic groove that recognizes client proteins, topped by an α-helical "lid" that regulates substrate access [126] [127]. These domains function independently yet communicate allosterically: ATP binding to the NBD induces conformational changes that open the SBD lid, facilitating substrate binding and release, while ATP hydrolysis stabilizes substrate interactions [126].
Cellular Localization and Transport Mechanisms
Hsp70 isoforms demonstrate specific subcellular localization patterns that dictate their functional specializations. Under normal conditions, Hsp70 predominantly resides in the cytoplasm, but stress conditions trigger rapid nuclear accumulation [19]. This translocation requires active transport mediated by specific nuclear transport receptors, particularly Hikeshi, which facilitates Hsp70's passage through nuclear pore complexes [19]. Mutational studies have identified three potential nuclear localization sequences (NLS) in human Hsp72: NLS1 (amino acids 245-264), NLS2 (568-574), and NLS3 (595-598) [19]. The functional significance of this compartmentalization is profound, as nuclear Hsp70 contributes to stress response coordination while cytoplasmic retention maintains its proteostatic functions.
Immunomodulatory Mechanisms of Hsp70
Pro-Inflammatory Functions: Immune Activation Pathways
Extracellular Hsp70 as a DAMP
When released into the extracellular space, Hsp70 functions as a damage-associated molecular pattern (DAMP) that activates innate immunity through pattern recognition receptors [126]. This extracellular Hsp70 (eHsp70) binds primarily to Toll-like receptors 2 and 4 (TLR2/4) on antigen-presenting cells including dendritic cells and macrophages [126] [128]. The binding triggers a MyD88-dependent signaling cascade that activates nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) [126]. This pathway subsequently induces the expression and secretion of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), thereby amplifying inflammatory responses and enhancing antigen-presenting cell activation [126].
Antigen Cross-Presentation Enhancement
Hsp70 significantly enhances adaptive immune activation through its specialized role in antigen cross-presentation. As a molecular chaperone, Hsp70 binds antigenic peptides and facilitates their delivery to major histocompatibility complex (MHC) class I molecules on antigen-presenting cells [129] [130]. This process enables cross-priming of CD8+ T cells, generating potent cytotoxic T lymphocyte (CTL) responses against chaperoned antigens [129]. The immunogenic potential of Hsp70-peptide complexes has been demonstrated in cancer vaccines, where tumor-derived Hsp70 complexes elicit robust anti-tumor immunity [130]. This unique capacity to channel bound peptides into the MHC I presentation pathway establishes Hsp70 as a critical bridge between innate and adaptive immunity.
Anti-Inflammatory Functions: Immune Suppression Pathways
Intracellular Hsp70-Mediated NF-κB Inhibition
In contrast to its extracellular pro-inflammatory effects, intracellular Hsp70 exerts potent immunosuppressive activity by directly inhibiting the NF-κB signaling pathway [127]. Mechanistically, Hsp70 overexpression blocks NF-κB activation and prevents nuclear translocation of the p50/p65 subunits through inhibition of IKK-mediated phosphorylation of IκB [127]. This suppression of NF-κB signaling downstream of various inflammatory receptors results in decreased production of pro-inflammatory cytokines and chemokines, effectively dampening immune activation at the transcriptional level [127].
Regulation of T Cell Differentiation
Hsp70 significantly influences adaptive immunity by modulating T helper cell differentiation平衡. Experimental evidence demonstrates that Hsp70 treatment increases Th17 frequencies and the Th17/Treg ratio while significantly decreasing Th1 cells and the Th1/Th2 ratio in human peripheral blood mononuclear cell cultures [131]. This skewing of T cell differentiation toward Th17 responses, coupled with suppression of Th1 responses, indicates Hsp70's capacity to reshape the adaptive immune landscape. Additionally, studies reveal that Hsp70 can promote regulatory T cell (Treg) differentiation under specific conditions, particularly in response to conserved Hsp70 peptides, suggesting context-dependent immunoregulatory functions [126] [127].
Table 1: Hsp70-Mediated Immune Modulation Pathways
| Immune Function | Mechanism | Signaling Pathways | Cellular Outcome |
|---|---|---|---|
| Pro-inflammatory | TLR2/4 activation | MyD88/NF-κB/MAPK | Dendritic cell maturation, pro-inflammatory cytokine release (TNF-α, IL-6, IL-1β) |
| Antigen Presentation | Peptide chaperoning | MHC I cross-presentation | CD8+ T cell activation, cytotoxic T lymphocyte responses |
| Anti-inflammatory | Intracellular NF-κB inhibition | IKK/IκB/NF-κB | Reduced pro-inflammatory cytokine production |
| T Cell Polarization | Th17/Treg modulation | Cytokine balance shift | Increased Th17/Treg ratio, decreased Th1/Th2 ratio |
Experimental Approaches for Investigating Hsp70 Immune Functions
Methodologies for Assessing Hsp70 Immune Activation
In Vitro Models of Extracellular Hsp70 Signaling
Research into Hsp70's pro-inflammatory functions employs well-established in vitro systems utilizing primary immune cells and cell lines. Experimental protocols typically involve treating human peripheral blood mononuclear cells (PBMCs), dendritic cells, or macrophages with highly pure, endotoxin-free recombinant Hsp70 (10-100 μg/mL) for 6-24 hours [131] [130]. Critical controls include endotoxin contamination checks using polymyxin B or LPS inhibitors, as endotoxin can confound results due to its high affinity for Hsp70 [130]. Readouts include flow cytometric analysis of surface activation markers (CD80, CD86, CD40, MHC II), multiplex cytokine assays measuring TNF-α, IL-6, IL-1β, and IL-10, and Western blotting or immunofluorescence for NF-κB nuclear translocation [126] [131]. Receptor specificity is validated using TLR2/TLR4 knockout cells or neutralizing antibodies [130].
Antigen Presentation Assays
The antigen cross-presentation capacity of Hsp70 is typically evaluated using ovalbumin (OVA) as a model antigen [129] [130]. Experimental workflows involve generating Hsp70-OVA complexes in vitro through coincubation (37°C for 30-60 minutes) followed by purification via size exclusion chromatography or ADP-agarose affinity columns [130]. These complexes are then pulsed into dendritic cells, with antigen presentation quantified using OVA-specific CD8+ T cells or T cell hybridomas that produce IL-2 upon recognizing OVA257-264 (SIINFEKL) peptide in context of H-2Kᵇ [129]. Intracellular antigen delivery can be tracked using fluorescently labeled OVA (e.g., AF647-OVA) and confocal microscopy [130].
Methodologies for Assessing Hsp70 Immunosuppression
Intracellular Signaling Inhibition Studies
Investigating Hsp70's anti-inflammatory functions requires models of intracellular Hsp70 overexpression. Common approaches include transfection with Hsp70 expression vectors, treatment with Hsp70 co-inducers (e.g., geranylgeranylacetone), or utilization of heat shock conditions (42-43°C for 30-60 minutes) to upregulate endogenous Hsp70 [127]. NF-κB inhibition is assessed by stimulating cells with TNF-α or LPS following Hsp70 induction, then measuring IκB phosphorylation/degradation, IKK activity, and NF-κB nuclear translocation via Western blotting, electromobility shift assays, or reporter gene assays [127]. Co-immunoprecipitation experiments validate direct interactions between Hsp70 and components of the NF-κB pathway [127].
T Cell Polarization Assays
Hsp70's impact on T helper cell differentiation is typically evaluated using naïve CD4+ T cells isolated from human PBMCs or mouse spleens [131]. Cells are activated with anti-CD3/anti-CD28 antibodies in the presence or absence of Hsp70 (5-20 μg/mL) under specific polarizing conditions: Th17 (TGF-β, IL-6, anti-IFN-γ, anti-IL-4), Th1 (IL-12, anti-IL-4), or Treg (TGF-β, anti-IFN-γ, anti-IL-4) [131]. After 3-5 days, polarized T cell populations are quantified using intracellular cytokine staining (IL-17A for Th17, IFN-γ for Th1) and flow cytometry, or FoxP3 staining for Tregs [131]. Supernatants are analyzed for characteristic cytokines (IL-17, IFN-γ, IL-10, IL-4) to confirm polarization efficiency [131].
Visualization of Hsp70 Signaling Pathways
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Hsp70 Immunomodulation Research
| Reagent/Category | Specific Examples | Research Application | Experimental Function |
|---|---|---|---|
| Recombinant Hsp70 | Endotoxin-free human/mouse Hsp70 (Abcam ab113187) | In vitro stimulation assays | Investigate extracellular Hsp70 effects on immune cells; typically used at 5-100 μg/mL |
| Hsp70 ELISA | HSP70 ELISA kit (Enzo ADI-EKS-715) | Quantitative measurement | Detect and quantify Hsp70 in serum, cell culture supernatants, or tissue lysates |
| Hsp70 Antibodies | FITC-conjugated anti-HSP70 (Abcam ab61907) | Flow cytometry, immunostaining | Intracellular Hsp70 detection in tissue digests or cultured cells |
| Genetic Models | HSP70 KO mice (MMRRC), TLR2/4 KO mice (Jackson Laboratory) | In vivo functional studies | Determine Hsp70-specific effects and receptor requirements in physiological contexts |
| Antigen Systems | Endotoxin-free ovalbumin (Invivogen), AF647-OVA (Thermo O34784) | Antigen presentation assays | Model antigen for tracking cross-presentation and intracellular delivery |
| Affinity Purification | ADP-Agarose (Sigma A2810) | Complex isolation | Purify functional Hsp70 or Hsp70-peptide complexes via nucleotide-dependent binding |
| Signaling Antibodies | Anti-MyD88 (R&D AF3109), Anti-TIRAP (Thermo PA5-88657) | Mechanism studies | Detect signaling pathway activation downstream of Hsp70 receptors |
Discussion: Therapeutic Implications and Future Directions
The dual immunomodulatory functions of Hsp70 present both challenges and opportunities for therapeutic development. In cancer contexts, extracellular Hsp70 demonstrates promising adjuvant properties for vaccine development by enhancing tumor antigen presentation and activating dendritic cells [19] [130]. Conversely, intracellular Hsp70 upregulation may benefit autoimmune disease treatment by suppressing NF-κB-mediated inflammation [127]. The context-dependent nature of Hsp70 immune functions necessitates precise targeting strategies, including isoform-specific inhibitors, co-chaperone disruption, and spatial delivery systems that distinguish between intracellular and extracellular pools.
Future research directions should prioritize understanding the molecular switches that determine Hsp70's immune activation versus suppression, particularly the role of post-translational modifications, co-chaperone interactions, and tissue-specific expression patterns. Additionally, advanced disease models recapitulating the complexity of Hsp70 networks in physiological environments will be essential for translating mechanistic insights into effective therapeutics. As part of the integrated chaperone system, Hsp70's relationship with Hsp60 and other chaperone families in maintaining proteostasis while directing immune outcomes represents a rich area for continued investigation with significant potential for clinical impact.
Cellular proteostasis is critically dependent on a network of molecular chaperones, with the 70 kDa heat shock protein (Hsp70) and chaperonin-containing TCP-1 (CCT, representing eukaryotic Hsp60) families serving as central components. These chaperone systems facilitate the folding of nascent polypeptides, prevent protein aggregation, refold misfolded proteins, and direct irreversibly damaged proteins for degradation [19] [132]. The Hsp70 system operates through an ATP-dependent cycle regulated by co-chaperones: Hsp40 (J-domain proteins) stimulates ATP hydrolysis, stabilizing Hsp70's interaction with client proteins, while nucleotide exchange factors (NEFs) promote ADP release, resetting the cycle [19]. CCT, a large multi-subunit complex, forms a folding chamber that encapsulates substrates, providing an isolated environment for folding [133]. In pathological conditions, particularly cancer and neurodegeneration, the chaperone system is frequently dysregulated. Cancer cells exhibit increased dependence on chaperones like Hsp70 and Hsp90 to stabilize oncoproteins and maintain survival under proteotoxic stress [19] [134]. This dependency has positioned chaperones as attractive therapeutic targets, leading to the development of both single-agent and combination targeting strategies.
Single-Agent Chaperone Targeting: Mechanisms and Limitations
Therapeutic Approaches and Efficacy
Single-agent chaperone inhibition primarily utilizes small molecules that disrupt ATP-binding or protein-protein interactions essential for chaperone function. For Hsp90, inhibitors like geldanamycin, 17-AAG (first-generation), ganetespib, luminespib (second-generation), and pimitespib (third-generation) target the N-terminal ATP-binding pocket, inhibiting the chaperone cycle and leading to proteasomal degradation of oncogenic client proteins [135]. Hsp70 targeting has proven more challenging due to its complex regulation and homology with constitutive Hsc70, though inhibitors like Pifithrin-μ show preclinical efficacy [134]. The table below summarizes representative single-agent chaperone inhibitors and their documented efficacy.
Table 1: Single-Agent Chaperone Inhibitors and Their Experimental Efficacy
| Target | Therapeutic Agent | Experimental Model | Reported Efficacy | Key Limitations |
|---|---|---|---|---|
| HSP90 | 17-AAG (Tanespimycin) | Glioma cell lines, orthotopic glioma mouse models [134] [135] | Arrested cell growth/proliferation, induced apoptosis, inhibited tumor growth in vivo [134] | Hepatotoxicity, low solubility, induction of heat shock response (HSR) [135] |
| HSP90 | Ganetespib | Various cancer models [135] | Potent in vitro activity, improved pharmacokinetics vs. 1st gen [135] | Gastrointestinal toxicity (diarrhea, nausea), elevated liver enzymes, limited clinical efficacy in monotherapy [135] |
| HSP90 | Pimitespib (TAS-116) | Clinical trials for various cancers [135] | Approved for monotherapy in some cancers; improved isoform selectivity [135] | Limited treatment responses in certain cancers, fatigue, elevated liver enzymes, mild GI toxicity [135] |
| HSP70 | Pifithrin-μ | GBM-bearing mice [134] | Inhibited tumor progression by activating pro-apoptotic UPR cascades [134] | The complex functional polymorphism of Hsp70 makes targeted inhibition challenging [19] |
| HSPA5/GRP78 | OSU-03012 | GBM cells and GBM-bearing mice [134] | Induced cell death, suppressed tumor growth, enhanced radiation efficacy [134] | Specificity and on-target toxicity concerns remain |
Mechanisms of Resistance to Single-Agent Therapy
Single-agent chaperone inhibitors face significant clinical challenges, primarily due to inherent and acquired resistance mechanisms. A fundamental limitation is the compensatory heat shock response (HSR), wherein inhibition of Hsp90 or Hsp70 activates heat shock factor 1 (HSF1), leading to transcriptional upregulation of HSP70, HSP27, and other cytoprotective chaperones. This creates a feedback loop that counteracts therapeutic efficacy and promotes cell survival [135]. Cancer cells also activate bypass signaling pathways; for example, following Hsp90 inhibition, PI3K/AKT and MAPK signaling pathways are often compensatory activated, maintaining survival signals despite degradation of specific oncogenic clients [135]. Furthermore, the redundancy and interconnectivity of the chaperone network means inhibiting one component (e.g., Hsp90) can be partially compensated by others (e.g., Hsp70), allowing client proteins to still achieve proper folding [19] [132]. These limitations have motivated the exploration of rational combination strategies.
Rationale and Mechanisms for Combination Chaperone Targeting
Combination strategies aim to achieve synergistic efficacy by concurrently targeting multiple nodes within the proteostasis network, overcoming the compensatory mechanisms that limit single-agent approaches. The core rationales include:
Vertical versus Horizontal Inhibition Strategies
- Vertical Inhibition: Targeting different components within the same chaperone pathway (e.g., simultaneously inhibiting Hsp90 and its co-chaperone CDC37) to create a more complete and synergistic blockade [6].
- Horizontal Inhibition: Targeting parallel proteostasis pathways (e.g., combining Hsp90 inhibition with proteasome or autophagy inhibition) to cripple complementary protein quality control systems and induce intolerable proteotoxic stress [132] [136].
Targeting Adaptive Stress Responses
Combination therapies can preemptively block the adaptive responses that cause resistance. For instance, co-targeting Hsp90 and HSF1 can prevent the compensatory upregulation of Hsp70 that normally blunts the effect of Hsp90 inhibitors [135].
Inducing Synthetic Lethality
In certain cancer contexts, simultaneous inhibition of two chaperone pathways is synthetically lethal, meaning the combination is highly toxic to cancer cells while sparing normal cells. This leverages the cancer cell's heightened dependence on proteostasis [134] [136].
The following diagram illustrates the core mechanistic rationale for combination targeting, highlighting how it disrupts multiple compensatory pathways.
Diagram 1: Mechanism of combination chaperone targeting. Single-agent inhibition triggers resistance via compensatory pathways, while combination strategies simultaneously block these adaptations to induce synergistic cell death.
Quantitative Efficacy Comparisons: Preclinical and Clinical Evidence
Direct comparisons of single-agent versus combination chaperone targeting reveal superior efficacy of combinatorial approaches across multiple disease models. The quantitative data summarized in the tables below demonstrate the enhanced therapeutic potential of these strategies.
Table 2: Preclinical Efficacy of Combination vs. Single-Agent Chaperone Targeting
| Combination Regimen | Experimental Model | Single-Agent Efficacy | Combination Efficacy | Proposed Mechanism |
|---|---|---|---|---|
| Proteasome Inhibitor (Bortezomib) + Autophagy Inhibitor (Lys05) [136] | Acute Myeloid Leukemia (AML) models | Limited efficacy due to backup autophagy activation [136] | Significantly reduced AML burden, extended survival, induced programmed cell death [136] | Concurrent blockade of two major degradation pathways, inducing terminal integrated stress response |
| HSP90 Inhibitor (NXD30001) + Radiotherapy [134] | GBM cells and mouse models | Inhibited tumor growth [134] | Increased radiosensitivity, enhanced tumor growth inhibition [134] | HSP90 inhibition destabilizes DNA repair clients, enhancing radiation-induced DNA damage |
| HSP90 Inhibitor (17-AAG) + Chemotherapy [134] [135] | Glioma cell lines and mouse models | Arrested cell growth [134] | Enhanced effects of chemo-radiotherapy [134] [135] | Degradation of oncogenic clients synergizes with DNA-damaging agents |
| HSP90 Inhibitor (XL-888) + Targeted Therapy [134] [135] | Neuroblastoma (NB) cell lines | Affected cancer-related processes (growth, apoptosis) [134] | Enhanced anti-tumor activity, overcoming resistance to targeted agents [135] | Prevents bypass signaling and resistance to kinase inhibitors |
Table 3: Clinical and Translational Evidence for Combination Chaperone Targeting
| Combination Strategy | Clinical/Translational Context | Key Findings | References |
|---|---|---|---|
| HSP90 Inhibitor + Immune Checkpoint Inhibitor (e.g., anti-PD-1/PD-L1) | Various solid tumors | Enhanced T cell activation and tumor immunogenicity; reversal of immunosuppressive tumor microenvironment. | [135] |
| Epichaperome Disruptor (PU-H71) as single agent | Cancer and Alzheimer's clinical evaluation | Targets disease-specific epichaperome scaffolds, restoring normal protein-protein interaction networks, not just inhibiting folding. | [72] |
| Proteostasis Network Disruption (Proteasome + Autophagy Inhibition) | Preclinical AML models (translational potential) | Significant sensitivity in primary AML cells vs. normal hematopoietic cells, suggesting a potential therapeutic window. | [136] |
Detailed Experimental Protocols for Key Combination Studies
Protocol: Evaluating Proteasome and Autophagy Dual Inhibition in AML
This protocol is based on the study that demonstrated effective dual targeting of proteostasis in acute myeloid leukemia [136].
Objective: To assess the synergistic effects of concurrently inhibiting the proteasome and autophagy pathways on AML cell viability and proteotoxic stress.
Materials and Reagents:
- Primary AML Cells: Isolated from patient samples or AML cell lines (e.g., MV4-11, MOLM-13).
- Healthy Donor Cells: Hematopoietic stem/progenitor cells (HSPCs) for toxicity comparison.
- Inhibitors: Proteasome inhibitor (Bortezomib, 10-20 nM), Autophagy inhibitor (Lys05, 1-10 µM).
- Cell Viability Assay: CellTiter-Glo Luminescent Cell Viability Assay.
- Apoptosis Detection: Annexin V-FITC / Propidium Iodide staining kit and flow cytometry.
- Protein Aggregation Staining: ProteoStat Aggresome Detection Kit.
- Western Blot Analysis: Antibodies for LC3-I/II, p62/SQSTM1, ubiquitin, CHOP, and cleaved caspase-3.
Methodology:
- Cell Culture and Treatment:
- Culture AML cells and normal HSPCs in appropriate media.
- Treat cells for 24-72 hours with: (a) Vehicle control (DMSO), (b) Bortezomib alone, (c) Lys05 alone, (d) Bortezomib + Lys05 combination.
Cell Viability and Synergy Assessment:
- Seed cells in 96-well plates and treat with serial dilutions of single agents and combinations.
- After 72 hours, measure viability using CellTiter-Glo assay.
- Analyze data using software like CalcuSyn to calculate Combination Index (CI) values. CI < 1 indicates synergy.
Apoptosis Analysis via Flow Cytometry:
- Harvest treated cells after 48 hours.
- Stain with Annexin V-FITC and PI according to manufacturer's protocol.
- Analyze on a flow cytometer. Quadrant analysis will distinguish early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cells.
Monitoring Proteotoxic Stress and Autophagy Flux:
- Protein Aggregation: Stain treated cells with ProteoStat dye and visualize by fluorescence microscopy/flow cytometry.
- Western Blot: Analyze lysates for markers of UPR (e.g., CHOP), autophagy flux (LC3-II accumulation, p62 degradation), and apoptosis (cleaved caspase-3).
In Vivo Efficacy Study (Xenograft Model):
- Engraft immunodeficient mice (NSG) with luciferase-labeled AML cells.
- Once engraftment is confirmed, randomize mice into four treatment groups: Vehicle, Bortezomib alone, Lys05 alone, Combination.
- Administer treatments intravenously or intraperitoneally on a defined schedule (e.g., twice weekly).
- Monitor tumor burden weekly via bioluminescent imaging.
- Record survival times and assess toxicity by monitoring body weight and organ histology.
Protocol: Assessing HSP90 Inhibitor and Radiotherapy Combination in Glioblastoma
This protocol outlines the methodology for evaluating the radiosensitizing effects of HSP90 inhibition in glioblastoma models [134].
Objective: To determine if HSP90 inhibition enhances the efficacy of radiotherapy in patient-derived glioblastoma (GBM) cells and orthotopic models.
Materials and Reagents:
- GBM Models: Patient-derived GBM stem-like cells (GSCs) and orthotopic mouse models.
- HSP90 Inhibitor: NXD30001 or other blood-brain barrier penetrant inhibitors (e.g., 17-AAG, AUY922).
- Irradiation Source: Preclinical X-ray irradiator.
- Clonogenic Assay Reagents: Crystal violet, methanol, acetic acid.
- DNA Damage Detection: Antibodies for γ-H2AX.
- Immunofluorescence: Standard reagents (PFA, Triton X-100, blocking serum, DAPI).
Methodology:
- In Vitro Radiosensitization Clonogenic Assay:
- Treat GSCs with HSP90 inhibitor or DMSO for 24 hours.
- Trypsinize, count, and seed appropriate cell numbers into 6-well plates.
- After 4-6 hours, irradiate plates with doses ranging from 0 to 8 Gy.
- Incubate cells for 10-14 days to allow colony formation.
- Fix and stain colonies with crystal violet. Count colonies (>50 cells) manually or with software.
- Plot survival curves and calculate the Dose Enhancement Ratio (DER).
DNA Damage Repair Analysis (γ-H2AX Foci):
- Culture GSCs on coverslips and pre-treat with HSP90 inhibitor for 24h.
- Irradiate with 2 Gy and fix cells at different time points post-IR (e.g., 0.5, 6, 24h).
- Perform immunofluorescence staining for γ-H2AX and counterstain nuclei with DAPI.
- Image using a fluorescence microscope and quantify γ-H2AX foci per nucleus. Delayed foci resolution in the combination group indicates impaired DNA repair.
In Vivo Orthotopic GBM Therapy Study:
- Stereotactically implant luciferase-expressing GSCs into the brains of immunodeficient mice.
- Monitor tumor establishment by bioluminescence imaging (BLI).
- Randomize mice into: (a) Vehicle, (b) HSP90 inhibitor alone, (c) Radiotherapy alone, (d) Combination.
- Administer HSP90 inhibitor via oral gavage. Deliver focal radiotherapy (e.g., 2 Gy fractions for 5 days) to the brain using a small animal irradiator.
- Monitor tumor growth weekly by BLI and record survival as the primary endpoint.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Chaperone Targeting Studies
| Reagent / Tool | Function / Application | Example Products / Citations |
|---|---|---|
| PU-H71 (Zelavespib) | Chemical probe; initially an HSP90 inhibitor, now known to selectively bind HSP90 incorporated into pathogenic epichaperomes, disrupting these scaffolds. | [72] |
| HSF1 Inhibitors | Suppresses the compensatory heat shock response (HSR) that is often upregulated in response to chaperone inhibition, preventing resistance. | Research compounds (e.g., KRIBB11, Rohinitib) |
| Lys05 | A potent autophagy inhibitor that blocks lysosomal acidification, used in combination with proteasome inhibitors to induce proteotoxic stress. | [136] |
| Bortezomib | Proteasome inhibitor that blocks the ubiquitin-proteasome system, leading to accumulation of misfolded proteins. | [136] |
| Pifithrin-μ | Inhibits Hsp70 function by blocking its interaction with co-chaperones and client proteins; used to study Hsp70's role in apoptosis and stress response. | [134] |
| Antibody Panel for UPR/HSR | Detect activation of proteostasis stress pathways via Western Blot or IHC. Key targets: HSP70, HSP27, HSF1, CHOP, XBP1s, LC3, p62. | Commercial vendors (CST, Abcam) |
| ProteoStat Aggresome Detection Kit | Fluorescent dye-based assay to detect and quantify protein aggregates in cells, a key readout for proteotoxic stress. | Enzo Life Sciences |
The comparative analysis of single-agent versus combination chaperone targeting unequivocally demonstrates the superior therapeutic efficacy of multi-target approaches. Single-agent inhibitors, while mechanistically insightful, are consistently limited by compensatory cellular responses, including heat shock factor activation, bypass signaling, and pathway redundancy. Combination strategies, which vertically or horizontally target the proteostasis network, induce synergistic proteotoxic stress that overwhelms cancer cell adaptability. The emerging concept of targeting epichaperomes—stable, disease-specific chaperone assemblies—represents a paradigm shift from inhibiting general chaperone function to dismantling pathological scaffolds [72]. Future directions will focus on the development of highly selective isoform-specific inhibitors, patient stratification based on proteostatic dependency biomarkers, and the rational design of multi-specific molecules that simultaneously engage multiple chaperone targets. Integrating chaperone modulators with immunotherapy, radiotherapy, and targeted agents holds significant promise for overcoming therapeutic resistance in oncology and neurodegenerative diseases, paving the way for more effective and personalized medicine.
Cellular protein homeostasis, or proteostasis, is a delicate balance between protein synthesis, folding, trafficking, and degradation, ensuring a stable and functional proteome [46]. The heat shock proteins Hsp70 and Hsp60 are central components of the proteostasis network, acting as molecular chaperones to prevent misfolding and aggregation, refold damaged proteins, and direct irreparably damaged proteins to degradation pathways [125]. In the context of neurodegenerative diseases, cancer, and other age-related disorders, this balance is disrupted, leading to a pathological state known as dysproteostasis [46]. Research into Hsp70 and Hsp60 mechanisms relies on a sophisticated toolkit of validation models, ranging from cellular assays that provide mechanistic insights to in vivo studies that offer a holistic view of chaperone function in physiology and disease. This technical guide synthesizes current methodologies and models for validating the functions and therapeutic targeting of these crucial chaperones, providing a framework for researchers and drug development professionals.
Hsp70 and Hsp60: Mechanisms and Functions
The Hsp70 Chaperone System
The Hsp70 family consists of multiple members located in various cellular compartments, including the cytoplasm, nucleus, endoplasmic reticulum (Hsp78/BiP), and mitochondria (Hsp75/mtHsp70) [125]. These chaperones function through an ATP-dependent cycle: Hsp70 binds to hydrophobic patches of unfolded client polypeptides, and ATP hydrolysis triggers a conformational change that stabilizes the client protein. The subsequent release of ADP and binding of a new ATP molecule discharges the folded client [137]. This cycle is critically regulated by co-chaperones, particularly Hsp40 (DNAJ) proteins, which deliver client proteins and stimulate Hsp70's ATPase activity [6] [125]. Hsp70's functions extend beyond folding to include protein translocation across membranes, dissolution of protein aggregates, and inhibition of apoptotic signaling [137] [125].
The Hsp60 Chaperonin System
Hsp60, also known as chaperonin 60 (Cpn60), primarily resides in mitochondria and forms a large double-ring tetradecameric structure that creates a central folding chamber [1]. As a Group I chaperonin, it functions in conjunction with a co-chaperone lid structure, Hsp10 (Cpn10). The folding process involves the encapsulation of non-native substrate proteins within the central cavity, where folding proceeds in an isolated environment, preventing aggregation [1]. In prokaryotes, this system exists as the well-characterized GroEL/GroES complex, while in eukaryotic mitochondria, it functions as mtHSP60/HSP10 [1]. Hsp60 is essential for the folding of numerous mitochondrial proteins and plays a critical role in maintaining mitochondrial proteome integrity, particularly under conditions of oxidative stress [1].
Table 1: Key Characteristics of Hsp70 and Hsp60 Chaperone Systems
| Feature | Hsp70 System | Hsp60 System |
|---|---|---|
| Primary Localization | Cytoplasm, nucleus, ER, mitochondria | Mitochondrial matrix |
| Structure | Monomer | Double-ring 14-mer (HSP60) + single-ring 7-mer (HSP10) |
| ATP Dependency | ATP-dependent | ATP-dependent |
| Key Co-chaperones | HSP40 (DNAJ), NEFs | HSP10 (CPN10) |
| Core Function | Polypeptide folding, translocation, disaggregation | Folding of naïve polypeptides in an enclosed chamber |
| Major Clients | Newly synthesized proteins, misfolded aggregates | Mitochondrial proteins |
Visualizing Chaperone Mechanisms
The diagram below illustrates the functional mechanisms of Hsp70 and Hsp60 chaperone systems in maintaining cellular proteostasis.
Cellular Assays for Chaperone Function Analysis
Intracellular Hsp70 Detection by Flow Cytometry
Flow cytometry provides a quantitative method for detecting stress-induced Hsp70 expression across multiple cell types, enabling rapid screening of compounds that modulate the heat shock response.
Protocol:
- Cell Preparation: Culture RAW264.7 macrophages, m-ICc12 intestinal crypt cells, primary bone marrow-derived dendritic cells (BMDC), or spleen cells in appropriate media [138].
- Stress Induction: Apply heat shock at 42.5°C for 1 hour or chemical stressor (e.g., sodium arsenite) with or without test compounds (e.g., carvacrol, curcumin, geldanamycin) [138].
- Recovery Phase: Incubate cells at 37°C for 4-18 hours to allow Hsp70 expression. Kinetics studies show Hsp70 levels peak between 4 hours and overnight recovery [138].
- Fixation and Permeabilization: Harvest cells, fix with 4% paraformaldehyde, and permeabilize with 0.1% Triton X-100 or commercial permeabilization buffers [138].
- Staining: Incubate with anti-Hsp70 primary antibody followed by fluorophore-conjugated secondary antibody. Include isotype controls for background subtraction [138].
- Analysis: Acquire data using flow cytometer and analyze mean fluorescence intensity (MFI) or percentage of Hsp70-positive cells. Compare stressed vs. unstressed conditions and compound-treated vs. untreated groups [138].
Hsp70-Specific T-Cell Activation Assay
This assay detects Hsp70 peptide presentation on MHC class II of antigen-presenting cells (APCs), relevant for immunomodulatory effects of chaperone targeting.
Protocol:
- Generate Hsp70-Specific T-Cell Hybridomas: Immunize mice with Hsp70-derived peptides and fuse responsive T cells with lymphoma partners to create stable hybridomas recognizing immunodominant Hsp70 peptides [138].
- APC Preparation and Treatment: Culture antigen-presenting cells (e.g., dendritic cells, macrophages) with test conditions (heat shock, compounds) [138].
- Co-culture: Incubate treated APCs with Hsp70-specific T-cell hybridomas for 24 hours [138].
- Activation Readout: Measure T-cell activation by ELISA detection of interleukin-2 (IL-2) or other cytokines in supernatant [138].
Hsp70 Activator Assessment in Prion Disease Models
Cellular models of protein misfolding evaluate the therapeutic potential of chaperone-targeting compounds like the Hsp70 activator SW02.
Protocol:
- Cell Culture: Maintain human neuroblastoma cells (SH-SY5Y) in DMEM-F12 medium with 10% FBS and 1% antibiotic cocktail at 37°C with 5% CO₂ [137].
- Neurotoxicity Induction: Treat cells with prion protein fragment PrP106-126 (100-200 μM) to model prion disease pathology. This peptide shares key properties with pathological PrPSc, including neurotoxicity and proteinase-K resistance [137].
- Compound Treatment: Apply Hsp70 activator SW02 (1-20 μM) 3 hours before PrP106-126 exposure. SW02 increases Hsp70 ATPase activity by approximately 45% [137].
- Viability Assessment: After 24 hours, measure cell viability using Cell Counting Kit-8 (CCK-8) or MTT assay. Calculate protection percentage relative to PrP106-126-only controls [137].
- Apoptosis Measurement: Analyze apoptosis by Annexin V/propidium iodide staining and flow cytometry, or caspase activity assays [137].
- Morphological Analysis: Assess neurite outgrowth and morphological changes via microscopy and image analysis software [137].
Table 2: Quantitative Results of Hsp70 Activator SW02 in Cellular Models
| Assay Type | Experimental Condition | Result | Significance |
|---|---|---|---|
| Hsp70 Expression | SW02 treatment | Increased Hsp70 mRNA expression | p < 0.01 |
| ATPase Activity | SW02 treatment | Enhanced Hsp70 ATPase activity | p < 0.01 |
| Cell Viability | PrP106-126 + SW02 pretreatment | Significant inhibition of cytotoxicity | p < 0.01 |
| Apoptosis | PrP106-126 + SW02 pretreatment | Significant reduction in apoptosis | p < 0.01 |
| Neurite Outgrowth | SW02 treatment | Promoted neurite extension | Qualitative improvement |
Reporter Assay for Transcriptional Activation
Reporter constructs measure activation of heat shock promoters, useful for high-throughput screening of Hsp inducers.
Protocol:
- Reporter Cell Line: Generate stable cell line with heat shock promoter (e.g., DNAJB1) driving luciferase expression [138].
- Treatment: Expose reporter cells to test compounds with or without stress conditions (e.g., arsenite) [138].
- Luciferase Measurement: Lyse cells after 6-24 hours and quantify luciferase activity using luminometer. Normalize to protein concentration or control vector [138].
Disease Organoids and 3D Models
While the search results do not provide specific protocols for Hsp70 and Hsp60 research in disease organoids, this emerging model system offers significant potential for chaperone research. Organoids—three-dimensional, self-organizing microtissues derived from stem cells—can recapitulate complex tissue architecture and cellular interactions not available in traditional 2D cultures. For neurodegenerative disease modeling, cerebral organoids containing neurons, astrocytes, and microglia could enable study of cell-non-autonomous chaperone functions and testing of chaperone-targeting therapeutics in a more physiologically relevant context [139].
In Vivo Validation Models
Prion Disease Mouse Model
In vivo models validate chaperone-targeting therapeutic potential in complex organismal contexts.
Protocol:
- Infection Model: Inoculate mice (e.g., C57BL/6) with ME7 scrapie strain via intracerebral or intraperitoneal injection [137].
- Compound Administration: Treat infected mice with Hsp70 activator SW02 via intraperitoneal injection. Begin treatment at various disease stages to assess preventive vs. therapeutic effects [137].
- Survival Monitoring: Monitor mice daily for survival, recording time to endpoint criteria (e.g., neurological signs, weight loss) [137].
- Tissue Analysis: At endpoint or predetermined timepoints, perfuse mice and collect brain tissue for analysis [137].
- Pathological Assessment:
- PrPSc Accumidation: Measure PrPSc levels in brain homogenates using proteinase K digestion and Western blotting or ELISA [137].
- Histopathology: Process brain sections for immunohistochemistry detecting GFAP (astrocytosis), Iba1 (microgliosis), and neuronal markers [137].
- Hsp70 Expression: Quantify Hsp70 levels in brain regions using Western blot, RT-qPCR, or immunohistochemistry [137].
Results: SW02 treatment in ME7 scrapie-infected mice extended survival from 217.6 ± 5.4 days (untreated) to 223.6 ± 6.0 days (p = 0.16) and significantly reduced PrPSc accumulation at 5 months post-injection (p < 0.05) without significant effect on GFAP expression [137].
Cancer Xenograft Models with HSP60 Expression
HSP60's role in cancer progression can be validated using xenograft models with correlation to clinical data.
Clinical Correlation Protocol:
- Patient Tissue Collection: Enroll cancer patients (e.g., ovarian cancer, n=260) and collect tumor tissues along with clinical data [140].
- Immunohistochemistry: Perform IHC staining for HSP60 on formalin-fixed, paraffin-embedded tissue sections [140].
- Scoring: Categorize patients into high- or low-HSP60 expression groups based on staining intensity and distribution [140].
- Data Analysis: Correlate HSP60 expression with clinicopathological features (tumor size, FIGO stage, lymph node metastasis) and survival outcomes (overall survival, disease-free survival) using Kaplan-Meier curves and Cox regression models [140].
Results: In ovarian cancer, high HSP60 expression associated with larger tumor size, advanced FIGO stage, increased lymph node metastasis, and significantly shorter overall and disease-free survival, identifying HSP60 as an independent prognostic factor [140].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Chaperone Validation Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cell Lines | SH-SY5Y (human neuroblastoma), RAW264.7 (mouse macrophage), Primary BMDCs | Cellular models for stress response, neurotoxicity, immunomodulation |
| Hsp70 Modulators | SW02 (activator), Curcumin (inducer), Geldanamycin (indirect inducer via Hsp90 inhibition) | Pharmacological manipulation of Hsp70 expression and function |
| Hsp60 Reagents | HSP60 antibodies for IHC/Western, HSP60 expression plasmids | Detection, localization, and overexpression studies |
| Detection Antibodies | Anti-Hsp70, Anti-Hsp60, Anti-GFAP, Anti-PrPSc | Protein quantification and localization in cellular and tissue samples |
| Assay Kits | Cell Counting Kit-8 (CCK-8), Annexin V Apoptosis Kit, IL-2 ELISA | Viability, apoptosis, and cytokine response measurements |
| Animal Models | ME7 scrapie-infected mice, Xenograft models, HSP60 transgenic mice | In vivo validation of therapeutic efficacy and biomarker correlation |
| Reporter Systems | DNAJB1-luciferase construct, HSE-driven fluorescent reporters | Monitoring heat shock pathway activation |
The comprehensive validation model framework presented—spanning cellular assays, emerging organoid technology, and in vivo studies—provides a rigorous approach for investigating Hsp70 and Hsp60 mechanisms in proteostasis. The integration of quantitative cellular screening with physiologically relevant animal models and clinical correlation creates a powerful pipeline for translating basic chaperone biology into therapeutic applications for neurodegenerative diseases, cancer, and other proteostasis-related disorders. As these models continue to evolve with technological advances, they will undoubtedly yield deeper insights into chaperone biology and enhanced opportunities for therapeutic intervention.
Conclusion
Hsp70 and Hsp60 represent central, yet functionally distinct, pillars of the cellular proteostasis network with profound implications for human health and disease. The structural and mechanistic insights into their ATP-dependent allosteric regulation provide a solid foundation for therapeutic intervention, while their integration into broader chaperone networks and the emerging concept of the epichaperome reveal new dimensions of complexity. Despite significant challenges in drug development—including functional redundancy, target selectivity, and compound delivery—recent advances in small-molecule inhibitors and protein-protein interaction disruptors show considerable promise. Future research directions should focus on isoform-specific targeting, combinatorial approaches that exploit chaperone network dependencies, and the development of biomarkers for patient stratification. The continued elucidation of chaperone mechanisms in specific pathological contexts will undoubtedly yield novel therapeutic strategies for cancer, neurodegenerative disorders, and other protein misfolding diseases, positioning molecular chaperones as critical targets in the next generation of precision medicine.
References
- 1. Molecular Chaperonin HSP60: Current Understanding and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121636/]
- 2. Genome-Wide Identification of HSP70 Gene Family and ... [https://www.mdpi.com/2311-7524/11/6/643]
- 3. The landscape of molecular chaperones across human ... [https://www.nature.com/articles/s41467-021-22369-9]
- 4. A quantitative chaperone interaction network reveals the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4104544/]
- 5. Mechanistic insights of molecular chaperone Hsp70 [https://www.sciencedirect.com/science/article/pii/S3050623925000513]
- 6. Advances in the structures, mechanisms and targeting of ... [https://www.nature.com/articles/s41392-025-02166-2]
- 7. Hsp70 molecular chaperones: multifunctional allosteric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7219557/]
- 8. Insights into the molecular mechanism of allostery in Hsp70s [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2015.00058/full]
- 9. Structural basis for active single and double ring ... [https://www.nature.com/articles/s41467-020-15698-8]
- 10. Oligomeric State and Holding Activity of Hsp60 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10177986/]
- 11. Pathways of allosteric regulation in Hsp70 chaperones [https://www.nature.com/articles/ncomms9308]
- 12. Allosteric Communication between the Nucleotide Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3138311/]
- 13. Crystal structures of dimeric and heptameric mtHsp60 ... [https://www.life-science-alliance.org/content/6/6/e202201753]
- 14. Molecular chaperones and neuronal proteostasis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4471145/]
- 15. Hsp70: A Multifunctional Chaperone in Maintaining ... [https://pubmed.ncbi.nlm.nih.gov/40214463/]
- 16. Navigating the landscape of protein folding and proteostasis [https://www.nature.com/articles/s41392-025-02439-w]
- 17. Heat shock proteins: Biological functions, pathological roles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9345296/]
- 18. Hsp70 chaperone: a master player in protein homeostasis. [https://f1000research.com/articles/7-1497]
- 19. Hsp70: A Multifunctional Chaperone in Maintaining ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989206/]
- 20. Conserved conformational selection mechanism of Hsp70 ... [https://elifesciences.org/articles/32764]
- 21. ATP Induced Conformational Changes in Hsp70 [https://pmc.ncbi.nlm.nih.gov/articles/PMC2787669/]
- 22. Dynamics, flexibility, and allostery in molecular chaperonins [https://www.sciencedirect.com/science/article/pii/S0014579315005529]
- 23. Hsp70 chaperones: Cellular functions and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2773841/]
- 24. Mechanisms and regulation of the Hsp70 chaperone network [https://www.nature.com/articles/s41580-025-00890-9]
- 25. Targeting Chaperone/Co-Chaperone Interactions with Small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8534281/]
- 26. Function, evolution, and structure of J-domain proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6363617/]
- 27. Chaperonin [https://en.wikipedia.org/wiki/Chaperonin]
- 28. Molecular chaperones of the Hsp110 family act as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1478182/]
- 29. Structural Basis for the Cooperation of Hsp70 and Hsp110 ... [https://www.sciencedirect.com/science/article/pii/S0092867408006788]
- 30. The Role of Co-chaperones in Synaptic Proteostasis and ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2017.00248/full]
- 31. The role of heat shock proteins in preventing amyloid toxicity [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1045616/full]
- 32. Molecular clamp mechanism of substrate binding by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC384753/]
- 33. Chaperone-mediated protein folding [https://pubmed.ncbi.nlm.nih.gov/10221986/]
- 34. Mechanism of Substrate Recognition by Hsp70 Chaperones [https://pubmed.ncbi.nlm.nih.gov/15270690/]
- 35. Visualizing chaperone-assisted protein folding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4937829/]
- 36. Short hydrophobic loop motifs in BRICHOS domains ... [https://www.nature.com/articles/s42003-023-04883-2]
- 37. Increased surface charge in the protein chaperone Spy ... [https://www.sciencedirect.com/science/article/pii/S0021925817497882]
- 38. Higher levels of heat shock proteins in longer-lived ... [https://www.sciencedirect.com/science/article/abs/pii/S0047637411000893]
- 39. "Compartmentalization - AP Biology Study Guide " [https://www.savemyexams.com/ap/biology/college-board/20/revision-notes/unit-2-cell-structure-and-function/cell--compartmentalization/compartmentalization-in-eukaryotic-cells/]
- 40. Mechanisms and regulation of the Hsp70 chaperone network [https://pubmed.ncbi.nlm.nih.gov/41145833/]
- 41. HSP70-Mediated Autophagy-Apoptosis-Inflammation Network ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12292420/]
- 42. HSP60 chaperone deficiency disrupts the mitochondrial ... [https://www.sciencedirect.com/science/article/pii/S2212877824001406]
- 43. Molecular mechanisms of mitochondrial quality control - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12400733/]
- 44. Mitochondrial quality control and transfer communication in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1542369/full]
- 45. T817MA attenuates oxidative stress and ER stress via the ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1701967/full]
- 46. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 47. How cryo‐electron microscopy and X‐ray crystallography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5192981/]
- 48. Comparing Analytical Techniques for Structural Biology [https://www.nanoimagingservices.com/about/blog/unraveling-the-mysteries-of-the-molecular-world-structural-biology-techniques]
- 49. Mechanisms of assembly and function of the Hsp70-Hsp40 ... [https://www.sciencedirect.com/science/article/abs/pii/S109727652500783X]
- 50. Revolutionary structures show how chaperone proteins ... [https://www.news-medical.net/news/20251015/Revolutionary-structures-show-how-chaperone-proteins-prevent-diseases-caused-by-protein-misfolding.aspx]
- 51. Hsp70: A Multifunctional Chaperone in Maintaining ... [https://www.mdpi.com/2073-4409/14/7/509]
- 52. Advancing time-resolved structural biology [https://www.nature.com/articles/s41592-025-02659-6]
- 53. Chaperone-client interactions: Non-specificity engenders ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5519353/]
- 54. Accelerating Drug Discovery With ATPase Activity Assays [https://bellbrooklabs.com/atpase-activity-assay-for-drug-discovery/?srsltid=AfmBOoo_DRvEeCnokZ-__-lpKStrs58AO4kAKBUeZJ08lj-rk-khv-U9]
- 55. Measuring In Vitro ATPase Activity for Enzymatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5091952/]
- 56. Distinct static and dynamic interactions control ATPase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2074893/]
- 57. Toward Developing Chemical Modulators of Hsp60 as ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2018.00035/full]
- 58. Functions and Therapeutic Potential of Extracellular Hsp60 ... [https://www.mdpi.com/2076-3417/11/2/736]
- 59. Targeting Allosteric Control Mechanisms in Heat Shock ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5502483/]
- 60. Emerging opportunities and challenges in small molecule ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425008657]
- 61. The Hsp70 chaperone system: distinct roles in erythrocyte ... [https://haematologica.org/article/view/haematol.2019.233056]
- 62. Molecular Chaperonin HSP60: Current Understanding and ... [https://www.mdpi.com/1422-0067/25/10/5483]
- 63. Discovery and Characterization of a Cryptic Secondary ... [https://pubmed.ncbi.nlm.nih.gov/35164081/]
- 64. Using Peptidomimetics and Constrained Peptides as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6017787/]
- 65. Recent advances in the development of protein– ... [https://www.nature.com/articles/s41392-020-00315-3]
- 66. Peptidomimetics: A Synthetic Tool for Inhibiting Protein– ... [https://link.springer.com/article/10.1007/s10989-019-09831-5]
- 67. Molecular mechanisms of chaperone‐directed protein folding [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895457/]
- 68. Chaperone (protein) [https://en.wikipedia.org/wiki/Chaperone_(protein)]
- 69. Heat shock proteins in protein folding and membrane ... [https://pubmed.ncbi.nlm.nih.gov/1680013/]
- 70. Peptides and peptidomimetics as regulators of protein-protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5496809/]
- 71. Heat shock proteins as hallmarks of cancer [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-024-01601-1]
- 72. redefining chaperone biology and therapeutic strategies in ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00010f]
- 73. More than Just Protein Folding: The Epichaperome ... [https://www.mdpi.com/2073-4409/14/3/204]
- 74. Epichaperomes: redefining chaperone biology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11933791/]
- 75. How Post-Translational Modifications Rewire Cellular ... [https://communities.springernature.com/posts/how-post-translational-modifications-rewire-cellular-networks-through-epichaperomes]
- 76. PTMs as molecular encoders: reprogramming chaperones ... [https://pubmed.ncbi.nlm.nih.gov/40877054/]
- 77. Article High-throughput screening identifies a novel small- ... [https://www.sciencedirect.com/science/article/abs/pii/S0026895X24230145]
- 78. High-throughput Screening Identifies Small Molecule ... [https://pubmed.ncbi.nlm.nih.gov/22920900/]
- 79. High-throughput screening [https://en.wikipedia.org/wiki/High-throughput_screening]
- 80. Quantitative high-throughput screening data analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4375054/]
- 81. High Throughput Screening (HTS) Services [https://www.evotec.com/solutions/drug-discovery-preclinical-development/discovery/hit-identification-services/high-throughput-screening-hts-services]
- 82. Pharmacological chaperones restore proteostasis of ... [https://www.sciencedirect.com/science/article/pii/S1043661824003013]
- 83. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target [https://pmc.ncbi.nlm.nih.gov/articles/PMC2895966/]
- 84. The role of heat shock proteins in tumorigenesis and their ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925011331]
- 85. Functional Analysis of Hsp70 Inhibitors - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3827032/]
- 86. Cancer cell responses to Hsp70 inhibitor JG-98 [https://www.nature.com/articles/s41598-017-14900-0]
- 87. Mapping the binding sites of challenging drug targets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9790766/]
- 88. Visualizing the chaperone-mediated folding trajectory of ... [https://www.sciencedirect.com/science/article/pii/S1097276523007955]
- 89. Coevolution-based prediction of key allosteric residues for ... [https://elifesciences.org/articles/81850]
- 90. Designing allosteric modulators to change GPCR G protein ... [https://www.nature.com/articles/s41586-025-09643-2]
- 91. HSP60 - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/hsp60]
- 92. Hsp60 Post-translational Modifications: Functional and ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2020.00095/full]
- 93. The Challenging Riddle about the Janus-Type Role of ... [https://www.mdpi.com/2076-3417/11/3/1175]
- 94. Human Hsp60 with Its Mitochondrial Import Signal Occurs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4022648/]
- 95. Report Compromised Mitochondrial Protein Import Acts as ... [https://www.sciencedirect.com/science/article/pii/S2211124719309532]
- 96. An Allosteric Inhibitor of Bacterial Hsp70 Chaperone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9124243/]
- 97. DnaK as Antibiotic Target: Hot Spot Residues Analysis for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0124563]
- 98. PES inhibits human-inducible Hsp70 by covalent targeting ... [https://www.sciencedirect.com/science/article/pii/S0021925820002069]
- 99. Differential Targeting of Hsp70 Heat Shock Proteins HSPA6 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5401876/]
- 100. The disruption of protein−protein interactions with co ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8245820/]
- 101. Combination Strategies with HSP90 Inhibitors in Cancer ... [https://www.mdpi.com/1424-8247/18/8/1083]
- 102. a selective strategy for targeting oncogenic kinases in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC12147015/]
- 103. Heat Shock Response and Heat Shock Proteins: Current ... [https://pubmed.ncbi.nlm.nih.gov/38673794/]
- 104. HSP90 inhibitors beyond oncology: optimization and ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625002272]
- 105. Hsp70 - a biomarker for tumor detection and monitoring of ... [https://ro-journal.biomedcentral.com/articles/10.1186/1748-717X-9-131]
- 106. Hsp70—A Universal Biomarker for Predicting Therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10452093/]
- 107. Evaluating Serum Heat Shock Protein Levels as Novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7564530/]
- 108. Role of Hsp70 in Post-Translational Protein Targeting [https://pmc.ncbi.nlm.nih.gov/articles/PMC9866221/]
- 109. Role of the Hsp90/Hsp70-Based Chaperone Machinery in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3046050/]
- 110. Mechanisms of the Hsp70 chaperone system - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5026485/]
- 111. Identification and characterization of molecular interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1276915/]
- 112. Functional Modules of the Proteostasis Network - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6942124/]
- 113. The proteostasis network and its decline in ageing [https://www.nature.com/articles/s41580-019-0101-y]
- 114. The Hsp70 and Hsp60 chaperone machines [https://pubmed.ncbi.nlm.nih.gov/9476895/]
- 115. Molecular chaperones in cellular protein folding [https://pubmed.ncbi.nlm.nih.gov/7773752/]
- 116. Human Proteostasis Network Annotation [https://www.proteostasisconsortium.com/pn-annotation/]
- 117. Nascent chains derived from a foldable protein sequence ... [https://www.nature.com/articles/s41598-024-61274-1]
- 118. The Impact of Hidden Structure on Aggregate Disassembly by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9302491/]
- 119. Mechanism of nascent chain removal by the ribosome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12215860/]
- 120. Article Proteasomes Activate Aggresome Disassembly and ... [https://www.sciencedirect.com/science/article/pii/S1097276513005893]
- 121. Impact of Heat Shock Proteins in Neurodegeneration [https://pmc.ncbi.nlm.nih.gov/articles/PMC9305912/]
- 122. The Interplay between Heat Shock Proteins and Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10854888/]
- 123. Heat shock proteins in oncology: Diagnostic biomarkers or ... [https://www.sciencedirect.com/science/article/abs/pii/S0304419X11000229]
- 124. Heat Shock Protein (HSP) Drug Discovery and Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC4995156/]
- 125. Modulation of Heat Shock Proteins Levels in Health and ... [https://www.mdpi.com/2073-4409/14/13/979]
- 126. The dual-function of HSP70 in immune response and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12034705/]
- 127. Heat Shock Protein 70 as a Double Agent Acting Inside ... [https://www.mdpi.com/1422-0067/21/15/5298]
- 128. The dual-function of HSP70 in immune response and ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1587414/full]
- 129. Dual roles of in situ generated HSP70 in antigen delivery and ... [https://pubmed.ncbi.nlm.nih.gov/41112303/]
- 130. Dual roles of in situ generated HSP70 in antigen delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528137/]
- 131. Autologous extracellular Hsp70 exerts a dual role in ... [https://www.sciencedirect.com/science/article/pii/S1355814523015559]
- 132. Targeting proteostasis for cancer therapy: current advances ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02472-x]
- 133. The structure of CCT–Hsc70NBD suggests a mechanism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4449276/]
- 134. Beneficial Handling of Molecular Chaperones ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468336/]
- 135. Combination Strategies with HSP90 Inhibitors in Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389308/]
- 136. New Combination Therapy Circumvents AML's Resistance ... [https://www.ajmc.com/view/new-combination-therapy-circumvents-aml-s-resistance-to-proteasome-inhibitors]
- 137. Assessment of the therapeutic potential of Hsp70 activator ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1411529/full]
- 138. Hsp70 expression and induction as a readout for detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2866976/]
- 139. Cellular Chaperones As Therapeutic Targets in ALS to ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2017.00251/full]
- 140. Identification and Clinical Validation of High HSP60 ... [https://pubmed.ncbi.nlm.nih.gov/39801927/]