Chaperone-Assisted Protein Refolding: Protocols, Mechanisms, and Clinical Applications
This article provides a comprehensive guide to chaperone-assisted protein refolding, bridging foundational principles with practical laboratory applications.
Chaperone-Assisted Protein Refolding: Protocols, Mechanisms, and Clinical Applications
Abstract
This article provides a comprehensive guide to chaperone-assisted protein refolding, bridging foundational principles with practical laboratory applications. It explores the fundamental mechanisms by which molecular chaperones, including Trigger Factor, Spy, and the GroEL/ES system, facilitate protein folding in vitro and in vivo. Detailed, actionable protocols for refolding model proteins like carbonic anhydrase B and slow-folding GFP are presented, alongside strategies for troubleshooting common aggregation issues. The content also covers advanced validation techniques such as Hydrogen–Deuterium Exchange Mass Spectrometry (HDX-MS) and the READ method for visualizing dynamic chaperone-substrate complexes. Finally, the article examines the critical translational role of chaperones in drug development and the targeting of protein misfolding diseases, making it an essential resource for researchers and scientists in biotechnology and biomedicine.
The Cellular Machinery of Folding: Understanding Chaperone Mechanisms
Protein folding, the process by which a linear amino acid chain attains its functional three-dimensional structure, represents one of the most fundamental challenges in molecular biology. The paradox posed by Cyrus Levinthal in 1969 highlighted the apparent impossibility of proteins exhaustively searching all possible conformations to find their native state within biologically relevant timescales [1] [2]. This Application Note examines the complementary mechanisms of spontaneous and chaperone-assisted protein folding, with particular emphasis on their roles in resolving Levinthal's paradox and their implications for experimental research and therapeutic development.
The "thermodynamic hypothesis," established by Anfinsen's pioneering experiments, demonstrated that the native structure of a protein is determined solely by its amino acid sequence and represents the most thermodynamically stable conformation under physiological conditions [3] [1]. However, the crowded intracellular environment presents additional challenges not present in vitro folding experiments, including the constant risk of aggregation and misfolding due to inappropriate interactions with other cellular components [4] [3].
Theoretical Framework: Solving Levinthal's Paradox
Energy Landscape Theory and Folding Funnels
Levinthal's paradox originates from the astronomical number of possible conformations available to a polypeptide chain. For a typical 100-residue protein, with each residue having at least three possible conformations, the chain could adopt up to 3¹⁰⁰ (~10⁴⁸) different structures. If the protein sampled conformations at picosecond rates, exhaustive search would require longer than the age of the universe, yet most small proteins fold spontaneously on millisecond to microsecond timescales [1] [2].
The resolution to this paradox lies in the concept of funnel-shaped energy landscapes, where proteins do not randomly sample all possible conformations but rather follow biased pathways toward the native state [1] [2]. In this framework, the folding process is guided by a gradual decrease in energy and conformational entropy as the protein approaches its native structure, effectively creating a "funnel" that directs the search process [1].
Table 1: Key Theoretical Concepts Resolving Levinthal's Paradox
| Concept | Explanation | Experimental Evidence |
|---|---|---|
| Funnel-shaped Energy Landscape | Rugged landscape with overall bias toward native state | Φ-value analysis, folding kinetics studies [1] |
| Nucleation-Condensation | Small native-like region serves as folding nucleus | Protein engineering experiments [3] |
| Foldon Assembly | Modular folding of independent structural units | Hydrogen-deuterium exchange [3] |
| Molten Globule Intermediate | Compact intermediate with secondary structure but flexible side chains | NMR, circular dichroism [4] |
In Vivo vs. In Vitro Folding: Fundamental Similarities
Recent experimental evidence suggests that for small single-domain water-soluble globular proteins, no fundamental difference exists between in vivo (co-translational) and in vitro refolding. NMR and FRET studies monitoring co-translational structure acquisition have demonstrated that polypeptides remain unstructured during elongation at the ribosome but fold into compact, native-like structures only when the entire domain sequence is available [4] [1].
For multi-domain proteins, however, sequential folding emerges as a crucial mechanism. The N-terminal domains of large proteins can fold before biosynthesis of the entire chain is complete, with domains folding vectorially as their nascent chain portions emerge from the ribosome [4]. This sequential accessibility may help explain how complex multi-domain proteins avoid the combinatorial explosion of possible conformations that Levinthal's paradox describes.
Spontaneous Protein Folding Mechanisms
Determinants of Spontaneous Folding
Spontaneous folding of single-domain globular proteins proceeds through a series of well-defined steps, beginning with the rapid formation of local secondary structures, followed by the acquisition of a molten globule intermediate, and culminating in the precise side-chain packing that characterizes the native state [4] [1]. The rate of this process exhibits a characteristic dependence on protein chain length, with smaller proteins folding more rapidly than larger ones [4].
The critical role of the folding nucleus—a specific set of native contacts that once formed accelerates the acquisition of remaining structure—has been demonstrated through Φ-value analysis, which identifies residues involved in the rate-limiting transition state [1]. This nucleation mechanism provides a structural explanation for how proteins navigate the folding landscape efficiently without exhaustive search.
Experimental Characterization of Spontaneous Folding
Table 2: Experimental Parameters for Spontaneous Folding of Model Proteins
| Protein | Chain Length | Folding Time | Thermodynamic Stability (ΔG) | Key Intermediate States |
|---|---|---|---|---|
| Small α-helical domains | 20-60 residues | Microseconds | 5-15 kcal/mol | Molten globule [5] |
| Medium mixed α/β proteins | 60-150 residues | Milliseconds | 7-12 kcal/mol | Pre-molten globule [4] |
| Large multi-domain proteins | 150-300 residues | Seconds to minutes | 10-20 kcal/mol | Domain-specific intermediates [4] |
Chaperone-Assisted Folding Mechanisms
The Cellular Folding Environment
Despite the inherent ability of many proteins to fold spontaneously, the crowded intracellular environment presents unique challenges that necessitate chaperone assistance. Molecular chaperones function primarily as aggregation inhibitors rather than catalysts of folding, binding to exposed hydrophobic surfaces on non-native proteins and preventing inappropriate interactions that could lead to misfolding or aggregation [4] [3].
The importance of chaperones becomes particularly evident under cellular stress conditions, where increased concentrations of unfolded proteins threaten proteostasis. Under such conditions, heat shock proteins (HSPs) are upregulated through the heat shock response (HSR) pathway, enhancing the cell's folding capacity and preventing toxic aggregation events [3].
Major Chaperone Systems and Mechanisms
Hsp70 Chaperone Cycle
The Hsp70 system represents one of the most extensively studied chaperone families. Recent molecular dynamics simulations have revealed that Hsp70 facilitates client protein folding through an ATP-dependent cycle involving open-lid and closed-lid states. The closed-lid state interacts with client proteins via specific conserved nonpolar residues, preventing nonnative hydrophobic collapse and allowing more efficient folding upon release [6].
Table 3: Major Chaperone Systems and Their Functions
| Chaperone System | Primary Cellular Role | Key Mechanism | Energy Source |
|---|---|---|---|
| Hsp70 | Co-translational folding, stress response | Lid closure prevents hydrophobic collapse | ATP hydrolysis [6] |
| GroEL/GroES | Folding of aggregation-prone proteins | Provides isolated folding compartment | ATP hydrolysis [4] |
| Hsp90 | Activation of signaling proteins | Conformational remodeling of clients | ATP hydrolysis [3] |
| Small HSPs | Stress-induced aggregation prevention | Formation of holding complexes | ATP-independent [3] |
GroEL/GroES Chaperonin System
The GroEL/GroES system provides a physically sequestered environment for protein folding, isolating vulnerable folding intermediates from the crowded cytosol. Contrary to earlier hypotheses suggesting GroEL might act as an "unfoldase," current evidence indicates it does not accelerate the overall folding process but rather serves as a transient trap that binds excess unfolded protein chains, thus preventing their irreversible aggregation [4]. This protective function is particularly critical for proteins with slow folding rates that would otherwise be vulnerable to aggregation during their extended folding time.
Experimental Approaches and Protocols
Quantifying Protein Stability and Folding Kinetics
Accurate measurement of protein stability parameters is essential for both basic research and therapeutic development. The folding free energy (ΔGfold) represents the key thermodynamic parameter describing the balance between folded and unfolded states, typically ranging from 5-15 kcal/mol for most functional proteins [5].
Diagram 1: Protein folding energy landscape showing the transition from unfolded to native state through a partially folded intermediate.
Key Methodologies for Folding Analysis
Equilibrium Unfolding Experiments
Protocol: Chemical Denaturation with Urea/Guanidine HCl
- Prepare protein samples (0.2-0.5 mg/mL) in denaturant solutions spanning 0-8 M urea or 0-6 M guanidine HCl
- Incubate for 2-4 hours at constant temperature to ensure equilibrium
- Monitor unfolding using far-UV circular dichroism (222 nm for α-helical content) or intrinsic fluorescence (tryptophan emission)
- Fit transition data to a two-state or multi-state model to extract Cm (midpoint denaturant concentration) and m-value (cooperativity parameter)
- Calculate ΔGfold using linear extrapolation method: ΔG = ΔG° - m[denaturant] [5]
Kinetic Folding Measurements
Protocol: Stopped-Flow Fluorescence
- Prepare native protein and denatured protein in 6 M guanidine HCl
- Rapidly mix denatured protein with refolding buffer (1:10 dilution) in stopped-flow apparatus
- Monitor fluorescence changes with time resolution of 1-1000 ms
- Fit resulting traces to single or multi-exponential functions to extract folding rates
- Perform experiments at multiple temperatures to obtain activation parameters [5]
Single-Molecule Force Spectroscopy
Protocol: AFM-based Protein Unfolding
- Immobilize protein constructs on gold surface via cysteine tags
- Approach surface with AFM tip and retract at constant velocity (100-1000 nm/s)
- Record force-extension curves showing characteristic unfolding peaks
- Analyze contour length increases to identify unfolded domains
- Determine unfolding forces and energies from multiple unfolding events [7]
Research Reagent Solutions
Table 4: Essential Research Reagents for Protein Folding Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Chemical Denaturants | Urea, Guanidine HCl | Equilibrium unfolding experiments [5] |
| Fluorescent Dyes | ANS, Sypro Orange | Molten globule detection, aggregation monitoring [4] |
| Proteostasis Regulators | Ver-155008 (Hsp70 inhibitor), PU-H71 (Hsp90 inhibitor) | Chaperone function studies [3] |
| Redox Buffers | GSH/GSSG, DTT | Disulfide bond formation studies [3] |
| Molecular Crowding Agents | Ficoll, Dextran | Mimicking intracellular environment [4] |
| Protease Inhibitors | PMSF, Protease inhibitor cocktails | Preventing proteolytic degradation during folding [5] |
Chaperone-Assisted Refolding Protocols
In Vitro Refolding with GroEL/GroES
Protocol: GroEL/GroES-Mediated Refolding
- Denature client protein in 6 M guanidine HCl for 2 hours
- Rapidly dilute denatured protein into refolding buffer containing GroEL (1:1 molar ratio)
- Incubate for 5 minutes to allow binding of unfolded chains to GroEL
- Add ATP (2 mM) and GroES (2:1 molar ratio to GroEL) to initiate folding
- Monitor refolding kinetics using appropriate spectroscopic methods
- Compare with spontaneous refolding in GroEL-free buffer to quantify chaperone effect [4]
Key parameters to optimize: GroEL-client ratio, ATP concentration, temperature, and refolding time. For aggregation-prone clients, the presence of GroEL/GroES typically increases functional yield by 3-10 fold compared to spontaneous refolding.
Hsp70-Assisted Folding Assay
Protocol: Analyzing Hsp70 Lid Closure Effects
- Prepare Hsp70 constructs (wild-type and lid mutants)
- Bind nucleotide-free Hsp70 to unfolded client protein (e.g., SH3 domain)
- Initiate folding by adding ATP and monitoring client conformation
- Use NMR restraints in molecular dynamics simulations to analyze folding pathways
- Compare folding efficiency between open-lid and closed-lid chaperone states [6]
This protocol has demonstrated that the closed-lid state of Hsp70 interacts with client proteins via specific conserved nonpolar residues, preventing nonnative hydrophobic collapse and enabling more efficient folding upon release.
Diagram 2: Hsp70 chaperone cycle showing ATP-dependent conformational changes that facilitate client protein folding.
Therapeutic Applications and Future Perspectives
The intricate relationship between spontaneous and chaperone-assisted folding has profound implications for human health and disease. Dysproteostasis—the collapse of proper protein homeostasis—is implicated in a growing list of human diseases, including neurodegenerative disorders, metabolic syndromes, and cancer [3]. In cancer cells, the proteostasis network is frequently reprogrammed to support rapid proliferation and survival under stress conditions, making chaperones attractive therapeutic targets.
Emerging technologies in the protein folding field include deep learning approaches for predicting folding pathways and stability effects of mutations [8], single-molecule techniques for observing real-time folding dynamics [5], and novel small molecule regulators of chaperone function [3]. The FiveFold methodology, which combines predictions from five complementary algorithms, represents a particularly promising approach for modeling conformational ensembles and capturing the dynamic nature of protein folding landscapes [8].
For researchers investigating protein refolding protocols, the experimental framework presented herein provides a foundation for developing optimized refolding strategies that leverage both spontaneous folding principles and chaperone-assisted mechanisms. As our understanding of the intricate interplay between these pathways deepens, so too will our ability to manipulate proteostasis for therapeutic benefit.
Molecular chaperones are highly conserved proteins critical for maintaining cellular protein homeostasis (proteostasis) by facilitating the folding, assembly, and disaggregation of proteins under both normal and stress conditions [9] [10]. They function as essential components of the protein quality control system, preventing the accumulation of misfolded and aggregated proteins associated with numerous diseases, including neurodegenerative disorders, cancer, and inflammatory conditions [9] [11] [10]. This application note details the experimental approaches for studying four major chaperone families—HSP70, HSP90, HSP60/Chaperonins, and small Heat Shock Proteins (sHSPs)—providing structured protocols and quantitative data to support research and drug discovery efforts.
The HSP70 Chaperone System
Functional Mechanism and Experimental Analysis
HSP70 chaperones operate through an ATP-dependent cycle of substrate binding and release, assisted by co-chaperones including J-domain proteins (JDPs) that target Hsp70s to substrates and nucleotide exchange factors (NEFs) that regulate complex lifetime [10]. The flexible lid domain undergoes conformational changes between open and closed states, which is crucial for client protein interaction [12] [6].
Table 1: Key Functional Parameters of HSP70 Chaperones
| Parameter | Experimental Value | Experimental Context |
|---|---|---|
| Client Protein Folding Efficiency | SH3 folds more effectively after sampling conformational space within closed-lid Hsp70 | NMR restraint-assisted MD simulations of nucleotide-free Hsp70 [12] [6] |
| Interaction Mechanism | Specific, highly conserved nonpolar residues prevent nonnative hydrophobic collapse | All-atom MD simulations with SH3 client [12] |
| Cellular Role | Assists de novo folding of 10-20% of bacterial proteins; higher percentage in eukaryotes | Cellular functional studies [10] |
Protocol: Analyzing HSP70 Client Folding Using Molecular Dynamics
Purpose: To investigate HSP70-assisted client protein folding through computational simulations. Applications: Mechanism of action studies, drug target identification, and mutation impact analysis.
Methodology:
- System Setup:
Simulation Execution:
Data Analysis:
- Quantify client protein folding efficiency by measuring native state formation probability.
- Analyze Hsp70-client interactions, focusing on specific conserved nonpolar residues [12].
- Determine the role of closed-lid state in preventing nonnative hydrophobic collapse of client upon chaperone release [12] [6].
The HSP90 Chaperone System
Functional Roles in Cellular Stress and Disease
HSP90 functions as a dimeric molecular chaperone complex that interacts with specific client proteins, particularly in signal transduction pathways. Unlike HSP70, HSP90 shows specialization for regulatory proteins including kinases, transcription factors, and steroid hormone receptors [10]. During chronic stress adaptation, Hsp90 couples cell size increase to augmented translation in a process termed the 'rewiring stress response', which is essential for cellular adaptation to prolonged mild stresses [13].
Table 2: HSP90 in Chronic Stress Adaptation
| Experimental Context | HSP90 Function | Biological Outcome |
|---|---|---|
| Chronic mild stress (up to 2 weeks) in mammalian cells | Essential for coupling cell size increase to augmented translation | Increased cellular resilience to persistent and subsequent stresses [13] |
| Adaptation to proteotoxic stress (e.g., heat) | Supports Hsf1-mediated 'rewiring stress response' | Adaptive cell size increase and total protein scaling [13] |
| Failure of HSP90 function | Impaired stress adaptation | Potential contribution to aging processes [13] |
The HSP60/Chaperonin System
Structural and Functional Characteristics
HSP60 (chaperonin 60) forms large double-ring barrel structures that provide a central cavity for protein folding. Group I chaperonins (HSP60) are found in eubacteria and eukaryotic organelles, while Group II chaperonins (TRiC/CCT) reside in the eukaryotic cytosol [9] [14]. Mammalian HSP60 exists primarily as a single heptameric ring that converts to a tetradecameric double-ring structure in the presence of ATP and forms a football-type complex with the co-chaperone HSP10 [15].
Unique Nucleotide Specificity of HSP60
Unlike bacterial GroEL, mammalian HSP60 exhibits dual nucleotide specificity with both ATPase and GTPase activities, suggesting distinct functional roles for these activities in chaperone function [15].
Table 3: Comparative Nucleotide Hydrolysis in HSP60
| Parameter | ATPase Activity | GTPase Activity |
|---|---|---|
| Allosteric Kinetics | Two apparent transitions with Hill coefficients 1.76 (first) and 5.52 (second) | Single sigmoidal curve with Hill coefficient ~2.3 [15] |
| HSP10 Effect | Suppressed hydrolysis activity | No suppression of hydrolysis [15] |
| Structural Outcome | Stable double-ring structure with HSP10 | Predominantly single-ring structures [15] |
| Functional Role | Productive refolding of denatured substrates | Supporting function in protein folding [15] |
Protocol: Assessing HSP60 Nucleotide-Dependent Oligomerization
Purpose: To characterize HSP60 oligomeric states and complex formation in response to different nucleotides. Applications: Chaperonin mechanism studies, nucleotide analog screening, and therapeutic development.
Methodology:
- Sample Preparation:
- Purify recombinant wild-type HSP60 (porcine cytosolic source described) [15].
- Prepare nucleotide solutions: ATP and GTP across concentration range (0-1.0 mM).
- Include HSP10 co-chaperone for complex formation studies.
Transmission Electron Microscopy:
- Incubate HSP60 (0.1 mg/mL) with nucleotides (ATP or GTP, 1 mM) with/without HSP10 for 10 minutes at 25°C [15].
- Apply 5 μL samples to carbon-coated grids, negative stain with 2% uranyl acetate.
- Acquire images using TEM at appropriate magnification (e.g., 50,000×).
Statistical Structural Analysis:
- Classify >100 molecules per condition into structural categories: single-ring (HSP60₇), double-ring (HSP60₁₄), bullet-type complex (HSP60₁₄-HSP10₇), football-type complex (HSP60₁₄-(HSP10₇)₂) [15].
- Calculate percentage distribution of oligomeric states for each condition.
- Expected outcome: ~70% double-ring structures with ATP+HSP10 vs. predominantly single-ring with GTP+HSP10 [15].
Small Heat Shock Proteins (sHSPs)
Characteristics and Neuroprotective Functions
Small heat shock proteins (12-43 kDa) constitute ATP-independent chaperones that prevent irreversible aggregation of denaturing proteins through holder chaperone activity [11] [16]. Humans encode ten sHSPs (HSPB1-HSPB10) with varying tissue distribution and expression patterns [11]. Recent research demonstrates that cytosolic sHSPs are imported into the mitochondrial intermembrane space (IMS) under basal conditions, where they operate as molecular chaperones [17].
Table 4: Small HSP Functions in Neurological Disorders
| sHSP | Alternative Name | Neurological Disease Associations | Regulation in Disease |
|---|---|---|---|
| HSPB1 | Hsp27 (human)/Hsp25 (mouse) | Amyotrophic lateral sclerosis, Alzheimer's, Parkinson's, Multiple Sclerosis | Upregulated [11] |
| HSPB5 | Alpha-B crystallin | Alexander's disease, Alzheimer's, Multiple Sclerosis, Parkinson's | Upregulated [11] |
| HSPB8 | Hsp22 | Amyotrophic lateral sclerosis, Alzheimer's, Charcot-Marie-Tooth disease | Variably regulated [11] |
Protocol: Mitochondrial Import Assay for sHSPs
Purpose: To investigate the subcellular localization and mitochondrial import of small heat shock proteins. Applications: Studying mitochondrial proteostasis, neurodegenerative disease mechanisms, and stress response pathways.
Methodology:
- Mitochondrial Isolation:
- Culture mammalian cells (HeLa, C2C12, or primary lymphoblasts) to 80-90% confluence.
- For stress conditions, apply heat shock (42-45°C for 20-60 minutes) then collect cells immediately.
- Isolate mitochondria using differential centrifugation with isotonic buffers [17].
Proteinase K Protection Assay:
- Divide mitochondrial samples into three aliquots:
- Intact mitochondria: No detergent
- Swollen mitochondria: + Hypo-osmotic buffer (e.g., 10 mM HEPES)
- Disrupted mitochondria: + Detergent (e.g., 1% Triton X-100)
- Treat all samples with proteinase K (50-100 μg/mL) for 15-30 minutes on ice.
- Stop reaction with PMSF (2 mM) and prepare samples for immunoblotting [17].
- Divide mitochondrial samples into three aliquots:
Analysis:
- Perform immunoblotting with antibodies against sHSPs (HSPB1, HSPB5, HSPB8).
- Include controls for mitochondrial compartments: TOM20 (outer membrane), TIMM23 (inner membrane), HSP60 (matrix).
- Interpretation: IMS-localized sHSPs protected in intact mitochondria but degraded upon swelling; matrix proteins remain protected until detergent treatment [17].
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Research Reagents for Chaperone Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Client Proteins | SH3 domain, misfolded luciferase, acid-denatured GFP | Substrates for folding and refolding assays [12] [15] |
| Nucleotide Analogs | ATPγS, GTPγS, AMP-PNP | Studying ATPase/GTPase cycles and conformational states [15] |
| Protease Assay Components | Trypsin, Proteinase K, PMSF | Protease sensitivity assays for complex formation [15] [17] |
| Structural Biology Tools | Negative stain EM, Crosslinkers, NMR restraints | Oligomeric state analysis and structural studies [12] [15] |
| Cell Culture Models | HeLa, C2C12, primary lymphoblasts | Mitochondrial import and stress response studies [17] |
The sophisticated experimental approaches detailed in this application note enable researchers to dissect the complex mechanisms of major chaperone families. From computational simulations of HSP70 conformational dynamics to biochemical assessments of HSP60 nucleotide specificity and mitochondrial import assays for sHSPs, these protocols provide robust methodologies for advancing our understanding of chaperone-assisted protein refolding. The growing evidence of chaperone involvement in neurodegenerative diseases, cancer, and aging highlights the importance of these systems as therapeutic targets, necessitating continued methodological development in the field.
Molecular chaperones constitute a diverse network of proteins essential for maintaining cellular proteostasis. They facilitate the proper folding, assembly, and localization of other proteins, preventing aggregation and mediating the recovery of misfolded states. Unlike enzymes with specific substrates, chaperones must recognize a vast array of client proteins primarily through exposed hydrophobic regions that are typically buried in the native state. The molecular mechanisms underlying this recognition—how chaperones identify their substrates, the kinetic pathways of binding, and the structural consequences for the client protein—form the foundation of chaperone biology. This application note explores the structural principles of chaperone-substrate interactions, detailing key experimental methodologies and providing quantitative insights into these fundamental processes. Understanding these mechanisms is critical for developing therapeutic interventions for diseases of protein misfolding and for improving recombinant protein production in biotechnology.
Fundamental Recognition Mechanisms
Key Biophysical Principles of Recognition
Chaperone-client interactions are characterized by several unifying biophysical principles that enable their broad specificity and functional efficacy.
- Weak, Hydrophobic Interactions: The binding interfaces between chaperones and their clients are typically characterized by weak affinity and transient interactions, predominantly mediated by hydrophobic contacts [18]. This low binding affinity is crucial as it allows the chaperone to recognize a wide range of sequences while permitting client release once folding is achieved.
- Hierarchy of Affinities: A key feature of productive chaperone function is the existence of a affinity hierarchy, where the chaperone has higher affinity for unfolded states than for the native, folded state of the client [18]. This ensures that folding, driven by the client's intrinsic hydrophobic collapse, readily outcompetes chaperone binding, facilitating release of the functional protein.
- Plastic Binding Grooves: Structural studies reveal that chaperone substrate-binding domains often display significant plasticity. For example, the substrate-binding groove of Hsp70 chaperones can accommodate extended conformations of various hydrophobic sequences, contributing to their promiscuity [19].
Predominant Pathway: Conformational Selection
A pivotal concept in understanding chaperone specificity is the mechanism by which the chaperone engages with its client. Recent structural and kinetic evidence strongly supports conformational selection as a primary mechanism for several major chaperone families.
- Hsp70 Conservation: Solution NMR studies quantifying the flux along binding pathways have established that both bacterial and human Hsp70 chaperones interact with clients by selectively binding the unfolded state from a pre-existing ensemble of interconverting structures [20]. The chaperone does not induce unfolding but rather selects for client conformations that are already unfolded or extended.
- Structural Basis for Selection: The substrate-binding domain (SBD) of Hsp70 contains a hydrophobic pocket that can only accommodate an extended polypeptide chain. This imposes a structural constraint that inherently selects against compact, folded structures [20].
- Functional Advantage: This mechanism allows Hsp70 to avoid inappropriate interactions with natively folded, functional proteins, thereby minimizing disruption to the cellular proteome while effectively targeting proteins in non-native states [20].
The following diagram illustrates the conformational selection mechanism used by Hsp70 chaperones.
Comparative Mechanisms Across Chaperone Classes
While conformational selection is a major mechanism, different chaperone classes employ variations on this theme, tailored to their specific cellular roles.
- Spy: A Permissive Scaffold: The bacterial chaperone Spy operates without ATP hydrolysis. It employs an amphiphilic, flexible binding surface that initially engages clients via electrostatic interactions, followed by stabilization through hydrophobic contacts [18]. Its concave binding surface allows the client to explore conformational space while protected from aggregation. Folding proceeds on the chaperone surface until the native state's hydrophobic core forms, destabilizing the chaperone-client complex and triggering release [18].
- Synthetic Nano-Chaperones (SNCPs): Bio-inspired porous nanoparticles have been engineered to mimic chaperone function. These SNCPs provide a tunable internal hydrophobic environment that stabilizes specific secondary structures, such as the α-helical conformation of a model p53 peptide [21]. The hydrophobicity of the internal surface directly influences the secondary structure of the encapsulated peptide, demonstrating how environmental polarity can guide folding.
Table 1: Key Structural Features in Chaperone-Substrate Recognition
| Chaperone | Recognition Mechanism | Binding Site Characteristics | Key Client Features Recognized |
|---|---|---|---|
| Hsp70 | Conformational Selection [20] | Hydrophobic groove in SBD; plastic surface [19] | Extended conformations with hydrophobic cores (Ile, Leu, Val) [20] |
| Spy | Hierarchical binding (electrostatic, then hydrophobic) [18] | Flexible, amphiphilic concave surface [18] | Unfolded states; promiscuous binding [18] |
| Synthetic Nano-Chaperone | Hydrophobic encapsulation [21] | Tunable internal hydrophobicity of porous nanoparticle [21] | Peptide sequence; stabilizes α-helical structures [21] |
Experimental Protocols for Studying Recognition
NMR Spectroscopy for Detecting Conformational Selection
Solution Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful technique for studying chaperone-client interactions at atomic resolution under physiological conditions. It allows researchers to distinguish between folded, unfolded, and chaperone-bound states of a protein and to quantify the kinetics of their interconversion [18] [20].
Protocol: Characterizing Hsp70-Substrate Interactions via Methyl TROSY
Sample Preparation:
- Isotopic Labeling: Produce (^{15})N-labeled or (^{13})C(^{1})H(^{3})-methyl-labeled (Ile, Leu, Val) client protein in E. coli. For larger chaperones like Hsp70, perdeuteration with specific methyl labeling is essential to reduce relaxation effects and enhance signal [20].
- Protein Purification: Purify chaperone (e.g., Hsp70) and client protein using standard affinity and size-exclusion chromatography.
- Buffer Conditions: Use an appropriate physiological buffer (e.g., 20-50 mM HEPES/KOH, pH 7.0-7.5, 50-150 mM KCl, 5-10 mM MgCl(_2)).
NMR Data Acquisition:
- Titration Experiment: Acquire a series of (^{1})H-(^{15})N Heteronuclear Single Quantum Coherence (HSQC) or methyl-TROSY spectra of the labeled client protein while titrating in unlabeled chaperone.
- Spectra Recording: Monitor chemical shift perturbations, line broadening, and the disappearance of cross-peaks corresponding to the free client as the chaperone is added. The appearance of new peaks indicates the bound state.
Data Analysis:
- Pathway Quantification: Analyze the NMR kinetic data to determine the flux along conformational selection versus induced fit pathways. This involves quantifying the populations of free and bound states and their exchange rates [20].
- Structural Insights: The specific perturbations observed reveal the client residues involved in binding and the degree of unfolding upon chaperone engagement.
Circular Dichroism (CD) for Monitoring Secondary Structure
Circular Dichroism (CD) Spectroscopy is used to monitor changes in the secondary structure of a client protein upon interaction with a chaperone or within a synthetic chaperone environment [21].
Protocol: Assessing Secondary Structure Changes in SNCPs
Sample Preparation:
- Encapsulation: Incubate the peptide (e.g., p53pep) with the synthetic nano-chaperone (SNCP) in aqueous buffer to form the SNCP-peptide complex [21].
- Control Samples: Prepare samples of the free peptide in aqueous solution and in a helix-inducing solution (e.g., 30% trifluoroethanol) for reference.
CD Measurement:
- Parameter Setup: Use a CD spectropolarimeter with a temperature-controlled cuvette. Standard parameters include a wavelength range of 190-260 nm, a bandwidth of 1 nm, and a path length of 0.1 cm.
- Data Collection: Record the CD spectrum of each sample at room temperature. For thermal stability, record spectra at increasing temperatures (e.g., from 25°C to 75°C).
Data Analysis:
- Spectral Interpretation: Analyze the resulting spectra for characteristic signatures: double negative minima at ~208 nm and ~222 nm indicate α-helical structure, while a single negative minimum near 218 nm suggests β-sheet, and a negative peak around 200 nm indicates random coil.
- Quantification of Helicity: Use algorithms like the CONTIN/LL program to deconvolute the spectra and estimate the relative percentage of different secondary structures [21].
The experimental workflow for analyzing chaperone-client interactions is summarized below.
Research Reagent Solutions
A successful research program in chaperone-substrate recognition relies on a toolkit of specialized reagents and materials. The following table details essential solutions for key experiments.
Table 2: Essential Research Reagents for Chaperone Recognition Studies
| Reagent / Material | Function / Application | Specific Example / Note |
|---|---|---|
| Isotope-labeled Amino Acids (e.g., (^{15})N, (^{13})C) | Enables NMR detection of protein structure and dynamics in solution. | (^{13})C(^{1})H(^{3})-methyl labeling of Isoleucine, Leucine, Valine for methyl-TROSY on large proteins [20]. |
| Refolding Buffers & Additives | Promote correct folding and prevent aggregation during refolding assays. | L-arginine (ArgHCl): Acts as an aggregation suppressor during dilution refolding [22]. Low denaturant concentrations (e.g., 0.5-1 M Urea): Can stabilize folding intermediates [22]. |
| Synthetic Nano-Chaperones (SNCPs) | Artificial system to study and control peptide folding; delivery vehicle. | Mesoporous Silica Nanoparticles with inner surface modified with octyl groups (SNCP8) to create a hydrophobic internal environment that stabilizes α-helical structure [21]. |
| Model Client Proteins | Well-characterized substrates for in vitro chaperone activity assays. | Unfolded proteins/peptides with hydrophobic stretches (e.g., sequences enriched in Ile, Leu, Val) for Hsp70 binding studies [19] [20]. |
| Nucleotide Analogs | Probing the ATP-dependent chaperone cycle (e.g., of Hsp70). | Non-hydrolyzable ATP analogs (e.g., ATPγS) to trap the ATP-bound state of Hsp70 and study its effect on substrate affinity [20]. |
Application Notes & Technical Data
Quantitative Insights into Binding and Refolding
The efficiency of chaperone-mediated recognition and refolding can be quantified through various parameters, providing a basis for optimizing experimental conditions.
Table 3: Quantitative Data on Chaperone Activity and Refolding
| Parameter / System | Reported Value / Efficiency | Experimental Context / Significance |
|---|---|---|
| Hsp70-Substrate Binding Affinity (Kd) | Weak, micromolar range (e.g., ~3 μM for unfolded Fyn SH3 bound to Spy) [18] | Weak affinity prevents kinetic trapping and allows for client release upon folding. |
| Spy: Folded vs. Unfolded Client Affinity | Kd (unfolded) ~3 μM; Kd (folded) ~50 μM [18] | Demonstrates the critical affinity hierarchy that drives the folding pathway. |
| Protein Refolding Yield (Dilution + Additives) | ≥80% recovery of active protein [22] | Significant improvement over conventional methods, highlighting the utility of additives like ArgHCl. |
| Protein Refolding Yield (Microfluidic Chip) | ≥70% recovery [22] | Rapid mixing and controlled diffusion reduce aggregation, enabling higher protein concentrations and shorter refolding times. |
| Chaperone Network Throughput | Mediates folding of ~62% of total cellular protein flux [23] | Underlines the massive scale and essential nature of chaperone function in proteome maintenance. |
Practical Considerations for Protocol Implementation
- Optimizing Refolding Conditions: When refolding proteins from inclusion bodies, the critical parameter is to minimize aggregation at intermediate denaturant concentrations. The use of a slow, graded dialysis or the incorporation of chemical additives like arginine can dramatically improve yields by suppressing off-pathway aggregation [22].
- Handling Synthetic Nano-Chaperones: For SNCPs, the hydrophobicity of the internal surface is a critical design parameter. Functionalization with longer aliphatic chains (e.g., octyl groups in SNCP8) creates a more hydrophobic environment, which was shown to favor the formation and stabilization of α-helical structure in a model peptide over β-sheet or random coil [21].
- NMR Sample Requirements: For studies aimed at distinguishing conformational selection from induced fit, high-quality, stable isotope-labeled samples are non-negotiable. The application of advanced NMR techniques like methyl TROSY is crucial for studying the large complexes formed by chaperones and their clients [20].
{article}
The Ribosome's Role: Trigger Factor and Cotranslational Folding Pathways
The ribosome is not merely a synthetic machine for polypeptide production but a central hub for coordinating the initial stages of protein folding. This process, known as cotranslational folding, is guided by the intricate architecture of the ribosome and facilitated by molecular chaperones, with Trigger Factor (TF) being the primary ribosome-associated chaperone in bacteria. TF docks near the polypeptide exit tunnel, creating a protected folding cradle where nascent chains can begin their structural organization while being shielded from the crowded cellular environment [24]. Recent advances reveal that the ribosome and its associated chaperones direct folding along pathways that are fundamentally distinct from refolding of full-length proteins in solution. This application note details the mechanisms of cotranslational folding, summarizes key quantitative findings, and provides protocols for studying these processes, providing a critical resource for researchers in protein science and therapeutic development.
Protein folding in vivo begins during synthesis on the ribosome, a vectorial process where the nascent chain (NC) emerges sequentially from the N- to C-terminus. Unlike refolding from denaturant, where the entire sequence is simultaneously available, cotranslational folding is constrained by the physical context of the ribosome. The narrow, negatively charged ribosome exit tunnel can accommodate α-helices but not larger tertiary structures, while the ribosome surface itself can thermodynamically destabilize folding domains [25]. This delay in folding can prevent unproductive interactions and misfolding, particularly in multi-domain proteins.
The first cellular component to encounter the majority of nascent polypeptides in bacteria is Trigger Factor. This highly abundant chaperone binds to the large ribosomal subunit near the exit tunnel and engages a wide spectrum of nascent chains [26] [24]. The functional cooperation between the ribosome and TF establishes a protected compartment—a molecular cradle—where nascent polypeptides can explore conformational space while being protected from proteolysis and aggregation [24]. Understanding the principles of this coordinated folding is essential for fundamental biology and has direct applications in mitigating aggregation-related diseases and optimizing the production of complex recombinant proteins.
Mechanism of Trigger Factor and Chaperone Coordination
Structural Basis of Trigger Factor Function
Trigger Factor is a 48-kDa modular protein consisting of three domains: an N-terminal ribosome-binding domain, a middle peptidyl-prolyl isomerase (PPIase) domain, and a C-terminal domain that serves as the primary nascent chain binding region [26]. The crystal structure of TF in complex with the ribosome reveals a unique "crouching dragon" conformation, where the chaperone arches over the exit site of the ribosomal tunnel [24]. This arrangement creates a protected, shielded space of approximately 55 x 55 Å, sufficient to accommodate folding intermediates of small protein domains.
The N-terminal domain of TF is both necessary and sufficient for ribosome binding, primarily interacting with ribosomal protein L23 [26] [24]. This docking position allows the C-terminal arms of TF to sample the emerging nascent chain. The affinity of TF for the ribosome is remarkably high, with a dissociation constant (KD) of approximately 1 μM, ensuring that TF is present in stoichiometric complexes with ribosomes under physiological conditions [26].
Multimodal Recognition of Nascent Chain Features
TF employs multiple mechanisms to interact with a diverse range of substrate proteins, demonstrating remarkable binding versatility:
- Recognition of Hydrophobic Regions: TF exhibits a strong preference for nascent chains exposing linear hydrophobic segments. The residence time of TF on a nascent chain correlates with the presence and exposure of these hydrophobic regions, with some interactions lasting up to 111±7 seconds [26].
- Interaction with Folded Domains: Surprisingly, TF can also bind folded domains, such as that of the ribosomal protein S7, indicating an alternative mode of action that may extend beyond chaperoning unfolded chains to include roles in ribosome assembly [26].
- Chaperone Coordination: TF acts as a gatekeeper at the ribosome exit site. Recent research demonstrates that TF binding is disfavored very close to the ribosome, allowing initial folding events to occur. TF recognizes compact folding intermediates that expose extensive unfolded surfaces and subsequently dictates access for the downstream chaperones DnaJ and DnaK [27]. This collaboration allows the chaperone network to protect incipient structure in the nascent polypeptide well beyond the ribosome exit tunnel.
Quantitative Data on Trigger Factor Interactions
Table 1: Quantitative Parameters of Trigger Factor Interactions with Nascent Chains and Ribosomes
| Parameter | Value | Experimental System | Significance |
|---|---|---|---|
| TF-Ribosome Binding KD | ~1 μM | E. coli [26] | High affinity ensures TF is ribosome-associated. |
| Cellular TF Concentration | ~50 μM | E. coli [26] | TF is in excess over ribosomes (~30 μM). |
| TF Residence Time on Ribosome | t½ ~ 10-14 s | In vitro reconstitution [26] | Rapid binding and release cycle at exit tunnel. |
| TF Residence Time on Nascent Chain | 111 ± 7 s (for Luciferase) | In vitro translation [26] | Much longer than ribosome binding, allows multiple TF binding events. |
| Chain Length for Domain Folding | ~330 amino acids (GEF-G domain) | Arrest Peptide Profiling [28] | Defines the point at which a complete domain is extruded and can fold. |
Table 2: Nascent Chain Features Influencing Trigger Factor Binding Affinity
| Nascent Chain Feature | Effect on TF Binding | Example Protein |
|---|---|---|
| Extended hydrophobic segments | Strong increase in binding affinity and residence time | Firefly Luciferase [26] |
| Lack of hydrophobic segments | Weak or transient interaction | Basic, hydrophilic proteins [26] |
| Signal sequences | Strong binding | pOmpA (secretory protein) [26] |
| Signal anchor sequences | No significant binding | FtsQ (membrane protein) [26] |
| Non-linear hydrophobic patches | Intermediate binding | Proteins with collapsed folding intermediates [26] |
Experimental Protocols and Methodologies
Protocol: Arrest Peptide Profiling for Cotranslational Folding
Principle: This high-throughput method uses a force-sensitive arrest peptide (AP) from the E. coli SecM protein to detect co-translational folding in live cells. Nascent chain folding generates mechanical force that pulls the AP from the ribosome exit tunnel, relieving translational arrest and enabling expression of a downstream reporter [28].
Procedure:
- Library Construction: Clone the gene of interest (GOI) fused in-frame to the SecM AP sequence and a fast-folding reporter (e.g., msGFP). Generate a truncated library of the GOI using time-dependent exonuclease digestion to create variants of different lengths.
- Dual Reporter System: Use a plasmid with two independent expression cassettes: one for the GOI-AP-msGFP fusion and another for an mCherry control to normalize for expression variation.
- Expression and Cell Sorting: Express the library in E. coli and analyze cells by fluorescence-activated cell sorting (FACS). Gate cells based on the log(IGFP/ImCherry) ratio, which reports on folding-induced arrest release.
- Deep Sequencing and Analysis: Isclude DNA from each sorted population and deep-sequence the 3'-ends of the library inserts. Calculate an "AP score" for each truncation variant based on its distribution across the sorting gates. A peak in the AP score profile indicates a co-translational folding event at a specific nascent chain length.
Applications: Delineating folding pathways of structurally intricate proteins, defining the exact chain length required for domain folding, and studying the impact of chaperone ablation on folding pathways with codon resolution [28].
Protocol: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) on Ribosome-Nascent Chain Complexes (RNCs)
Principle: HDX-MS measures the exchange of backbone amide hydrogens with deuterium in the solvent. The rate of exchange is exquisitely sensitive to protein conformation, with structured regions exchanging more slowly. This allows probing the local folding status of a nascent chain on the ribosome at peptide-level resolution [25].
Procedure:
- RNC Preparation: Engineer and express DHFR (or protein of interest) constructs truncated at specific codon positions, followed by a strong ribosome stall sequence. Purify stable RNCs from E. coli using sucrose cushion centrifugation and size-exclusion chromatography.
- Deuterium Labeling: Expose purified RNCs to deuterated buffer for defined time points (e.g., 10 s, 100 s, 1000 s). Quench the reaction with low pH and low temperature.
- Proteolysis and LC-MS/MS Analysis: Digest the entire sample (including ribosomal proteins and chaperones) with an acid-tolerant protease (e.g., pepsin). Separate the resulting peptides using liquid chromatography and analyze by mass spectrometry.
- Data Processing: Identify peptides and calculate their deuterium uptake. Compare the deuteration patterns of nascent chain peptides across different RNC lengths and to a fully folded, released control.
Applications: Revealing non-native folding intermediates on the ribosome, mapping the path of the emerging polypeptide, and studying the structural impact of chaperones like TF on nascent chains without disrupting the complex [25].
Visualization of Experimental Workflows
Arrest Peptide Profiling Workflow
Diagram Title: Arrest Peptide Profiling for Cotranslational Folding Analysis
HDX-MS Workflow for RNC Analysis
Diagram Title: HDX-MS Workflow for Ribosome-Nascent Chain Complexes
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Studying Cotranslational Folding
| Reagent / Material | Function / Application | Key Features & Considerations |
|---|---|---|
| Trigger Factor Variants (e.g., Cysteine mutants for labeling) | Site-specific labeling with fluorophores (e.g., NBD, BADAN) to monitor conformational changes and binding kinetics. | Select mutants (e.g., TF14, TF150, TF326, TF376) for labeling different domains [26]. |
| Ribosome-Nascent Chain Complexes (RNCs) | Defined intermediates for biochemical and biophysical studies (e.g., HDX-MS, fluorescence spectroscopy). | Use stall sequences (e.g., SecM variants) for efficient and stable RNC purification [25]. |
| Fluorescent Dyes (NBD, BADAN, ANS) | Environmentally sensitive probes for monitoring substrate binding and conformational changes in TF and nascent chains. | NBD fluorescence increases in hydrophobic environments, reporting on TF-substrate interaction [26]. |
| Arrest Peptide (AP) Reporter Plasmids | High-throughput detection of co-translational folding forces in live cells (AP Profiling). | Ensure constructs contain SecM AP, a candidate protein, and a rapidly folding reporter (e.g., msGFP) [28]. |
| Deuterated Buffers | Solvent for HDX-MS experiments to measure hydrogen-deuterium exchange in proteins and complexes. | Critical for quantifying backbone amide protection and thus protein conformation and dynamics [25]. |
The ribosome provides a specialized platform that actively shapes the folding landscape of nascent proteins. Through its physical constraints and partnership with a dedicated chaperone network led by Trigger Factor, it ensures that the complex process of protein maturation proceeds with high fidelity and efficiency. The experimental approaches detailed here—from high-throughput in vivo profiling to peptide-resolution structural proteomics—provide a powerful toolkit for dissecting these pathways. As these methods continue to evolve, they will yield deeper insights into protein homeostasis, with broad implications for understanding disease mechanisms and developing novel biotherapeutic strategies.
{/article}
Cellular protein homeostasis, or proteostasis, is fundamental to cellular health and functionality, ensuring a stable and functional proteome capable of executing essential life tasks [29]. Molecular chaperones are pivotal guardians of the proteome, assisting in protein folding, preventing aberrant protein aggregation, and maintaining proteostasis balance [30] [29]. These chaperones can be broadly classified into two major categories based on their energy requirements: ATP-dependent chaperones, which consume ATP to fuel cycles of substrate binding, folding, and release, and ATP-independent chaperones, which operate without energy consumption, primarily preventing aggregation through stable substrate binding [31] [30] [32]. Understanding the distinct mechanisms, structures, and functional roles of these systems is crucial for developing targeted therapeutic strategies for diseases linked to proteostasis collapse, such as neurodegenerative disorders and cancer [29] [33]. This note details the core principles, experimental methodologies, and key reagents for studying these essential cellular components.
Core Principles and Key Distinctions
ATP-Dependent Chaperone Systems
ATP-dependent chaperones utilize ATP binding and hydrolysis to power conformational changes that drive iterative cycles of client protein binding, folding, and release [31] [33]. They often act as "foldases" by directly facilitating the refolding of non-native substrates [32].
Table 1: Major ATP-Dependent Chaperone Families and Their Functions
| Chaperone Family | Key Representatives | ATPase Cycle Features | Primary Cellular Functions |
|---|---|---|---|
| Hsp70 System | DnaK (prokaryotic), Hsp70 (eukaryotic) | Cycle regulated by Hsp40 (J-protein) and NEFs (e.g., GrpE) [31]. | Stabilization of nascent chains, prevention of aggregation, translocation across membranes [31]. |
| Hsp90 System | Hsp90α, Hsp90β, GRP94, TRAP1 [33] | Conformational cycle between open and closed states; regulated by co-chaperones (p23, Aha1) [31] [33]. | Maturation of specific "client" proteins like transcription factors and signaling kinases [31] [33]. |
| Chaperonins (Hsp60) | GroEL (bacteria), HSP60 (eukaryotes) | Folding occurs in an isolated Anfinsen cage upon GroES binding and ATP hydrolysis [31]. | Folding of proteins with complex α/β topologies; forceful unfolding and encapsulation of substrates [31]. |
| Hsp100 Unfoldases/Disaggregases | ClpB, Hsp104, ClpX | Hexameric AAA+ ATPases; sequential or concerted ATP hydrolysis [31]. | Protein disaggregation (with Hsp70), unfolding and translocation to proteases (e.g., ClpP) [31]. |
ATP-Independent Chaperone Systems
ATP-independent chaperones function without energy consumption, primarily acting as "holdases" that rapidly bind to non-native proteins to prevent their aggregation, especially under stress conditions [30] [32]. They are crucial under ATP-depleting stress conditions (e.g., oxidative stress) and in cellular compartments with limited ATP availability, such as the bacterial periplasm [32]. Notably, recent research has revealed that some ATP-independent chaperones, such as Spy, can also exhibit "foldase" activity, allowing certain substrates to reach their native state while still chaperone-bound [34].
Table 2: Characteristics of Select ATP-Independent Chaperones
| Chaperone | Class / Size | Activation Mechanism | Mode of Action / Specificity |
|---|---|---|---|
| Small Heat Shock Proteins (sHSPs) | e.g., HSPB1 (HSP27), HSPB5; 12-43 kDa oligomers [33] | Oligomeric rearrangement and increased substrate affinity upon stress [33] [32]. | Holdase; broad substrate specificity; first line of defense against aggregation [33] [32]. |
| Spy | Periplasmic chaperone [34] | Conformational dynamics fine-tune substrate affinity [32]. | Substrate-specific action (holdase or foldase) dictated by affinity for non-native states [34]. |
| Trigger Factor (TF) | Ribosome-associated chaperone [25] | Binds ribosome and engages nascent chains co-translationally [25]. | Holdase/foldase; prevents aggregation of nascent polypeptides; can allow folding while bound [25]. |
| HdeA/HdeB | Periplasmic, acid-activated [32] | Conformational activation and dimer dissociation at low pH [32]. | Holdase; prevents aggregation in the acidic environment of the stomach [32]. |
The diagram below illustrates the fundamental operational cycles of these two chaperone systems.
Experimental Protocols for Chaperone Mechanism Analysis
Protocol: Analyzing Cotranslational Folding with HDX-MS
This protocol, adapted from studies on E. coli dihydrofolate reductase (DHFR), details how to resolve chaperone-assisted protein folding on the ribosome at peptide resolution using Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) [25].
1. Preparation of Stalled Ribosome-Nascent Chain Complexes (RNCs): - Cloning: Generate DNA constructs encoding the protein of interest (e.g., DHFR) truncated at specific lengths, fused C-terminally to a strong ribosome stall sequence (e.g., an 8-amino acid stalling sequence) [25]. - Expression and Purification: Express the constructs in an appropriate bacterial strain (e.g., E. coli). Purify stalled RNCs using sucrose density gradient centrifugation or affinity-based methods. Verify stalling and homogeneity by checking puromycin insensitivity and via SDS-PAGE and mass spectrometry analysis [25].
2. Hydrogen-Deuterium Exchange (HDX) Labeling: - Deuteration: Initiate the exchange reaction by diluting the purified RNCs into deuterated buffer (e.g., D₂O-based refolding buffer). Perform labeling for a series of time points (e.g., 10 s, 100 s, 1000 s) at a controlled temperature (e.g., 25°C) to capture different exchange kinetics [25]. - Quenching: Stop the exchange reaction by lowering the pH and temperature. A typical quenching solution is ice-cold buffer at pH ~2.5 [25].
3. Sample Processing and Mass Spectrometry Analysis: - Digestion: Rapidly digest the quenched sample with an immobilized acid-tolerant protease (e.g., pepsin) [25]. - LC-MS/MS Analysis: Separate the resulting peptides using liquid chromatography under quenched conditions. Analyze the peptides using high-resolution mass spectrometry to determine the mass increase due to deuterium incorporation for each peptide [25].
4. Data Interpretation: - Mapping Protection: Compare the deuterium uptake of peptides from the nascent chain across different RNC lengths and labeling times. Peptides involved in stable structure or protected by chaperone binding will show reduced deuterium uptake compared to unstructured regions [25]. - Pathway Definition: Identify the order of structure formation by observing which regions become protected from exchange at specific nascent chain lengths, thereby defining the cotranslational folding pathway and the impact of associated chaperones like Trigger Factor [25].
Protocol: Assessing Chaperone Activity via Aggregation Prevention
This protocol measures the ability of a chaperone to prevent the aggregation of a model substrate under stress conditions (e.g., heat or chemical denaturation) and is applicable to both ATP-dependent and ATP-independent chaperones [32] [34].
1. Experimental Setup: - Reagents: Prepare assay buffer, the chaperone of interest, and a model substrate protein known to aggregate upon thermal or chemical denaturation (e.g., citrate synthase, insulin) [32] [34]. - Instrumentation: Use a spectrophotometer or fluorometer equipped with a temperature-controlled cuvette holder and a stirrer. Light scattering at 360 nm or an increase in fluorescence of dyes like Thioflavin T can monitor aggregation [32].
2. Aggregation Assay: - Control Reaction: In a cuvette, add the substrate protein to the assay buffer pre-equilibrated at the stress-inducing temperature (e.g., 45°C) or containing a denaturant. Initiate aggregation and record the increase in light scattering over time [32]. - Chaperone Reaction: Repeat the measurement in the presence of the chaperone. Pre-incubate the chaperone with the substrate or add them simultaneously to the stress conditions [32]. - For ATP-Dependent Chaperones: Include an ATP-regenerating system in the reaction buffer to sustain the ATPase cycle [31].
3. Data Analysis: - Initial Rate: Calculate the initial rate of aggregation from the steepest slope of the scattering curve. - Lag Time: Measure the time before a rapid increase in scattering occurs. - Final Extent: Compare the maximum scattering signal achieved in the control versus the chaperone-containing reaction. Effective chaperones will significantly reduce the initial rate, extend the lag time, and decrease the final extent of aggregation [32] [34].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Chaperone Research
| Reagent / Material | Function in Experimentation | Example Application |
|---|---|---|
| Stalled Ribosome-Nascent Chain Complexes (RNCs) | To capture and study defined intermediates during co-translational folding [25]. | Mapping folding pathways of nascent chains with HDX-MS [25]. |
| ATP-Regenerating System | To maintain constant ATP levels for sustained activity of ATP-dependent chaperones during long assays [31]. | Hsp70/Hsp40 refolding assays; GroEL/GroES encapsulation experiments [31]. |
| Model Aggregation-Prone Substrates | Client proteins used to quantitatively assess chaperone "holdase" activity [32] [34]. | Citrate synthase, insulin, and apoflavodoxin for thermal or chemical denaturation assays [32] [34]. |
| Site-Directed Mutagenesis Kits | To generate chaperone mutants for probing structure-function relationships and mechanistic studies [34]. | Creating affinity variants of Spy to study the relationship between binding strength and mechanism [34]. |
| Deuterium Oxide (D₂O) & Quenching Buffers | Essential components for HDX-MS experiments to monitor protein dynamics and folding [25]. | Probing the conformational status and protection of nascent chains on ribosomes [25]. |
Visualization of a Chaperone-Assisted Folding Pathway
The following diagram details the specific pathway of co-translational folding for a model protein like DHFR, as resolved by HDX-MS, highlighting the roles of the ribosome and chaperones.
Practical Refolding Protocols: From Denaturant to Native State
The production of recombinant proteins in bacterial systems like Escherichia coli frequently results in the formation of inclusion bodies (IBs)—protein aggregates with non-native conformations that lack biological activity [22] [35]. While containing relatively pure and intact recombinant proteins, these aggregates require refolding to recover active, correctly folded proteins suitable for research, diagnostics, and therapeutic development [22] [36]. The core challenge of in vitro refolding lies in managing the kinetic competition between first-order folding reactions and second-or higher-order aggregation pathways, the latter being favored at higher protein concentrations [37]. This application note details three fundamental refolding techniques—dilution, dialysis, and on-column refolding—within the context of modern chaperone-assisted research, providing structured protocols and quantitative comparisons to guide researchers in reconstituting biologically active proteins from inclusion bodies.
Core Refolding Principles and the Kinetic Competition
Protein refolding from a denatured state is a self-assembly process driven by intramolecular interactions. However, during refolding, transient folding intermediates expose hydrophobic regions that can interact intermolecularly, leading to irreversible aggregation [38] [35]. This aggregation follows higher-order kinetics than folding, making it particularly dominant at high protein concentrations [35] [37]. The primary goal of any refolding technique is to navigate the protein through this aggregation-prone window while maintaining the protein at the highest possible concentration to maximize the yield of the native fold [35]. The presence of disulfide bonds adds complexity, requiring carefully controlled redox conditions to facilitate correct pair formation [36] [39].
Table 1: Key Performance Indicators of Major Refolding Techniques
| Technique | Typical Recovery Yield | Typical Protein Concentration | Time Requirement | Key Advantage | Primary Limitation |
|---|---|---|---|---|---|
| Direct Dilution | ≤40% [22] | 1–10 mg/mL [22] | 1-2 days [22] | Simplicity of operation | Large volume, low final concentration |
| Dilution with Additives | ≥80% [22] | 1–10 mg/mL [22] | 1-2 days [22] | High refolding yield | Requires additive optimization |
| Dialysis | ≤40% [22] | 1–10 mg/mL [22] | 1-2 days [22] | Constant protein concentration | Slow, aggregation at medium denaturant |
| On-Column Refolding | Varies by protein [36] | Limited by column capacity | Several hours [36] | Reduced aggregation, simple buffer exchange | Requires His-tag, optimization |
| Microfluidic Chip | ≥70% [22] | ≥250 μg/mL [22] | ≥10 min [22] | Rapid mixing, controllable gradient | Specialized equipment needed |
Established Refolding Methodologies
Direct Dilution
Principle
The direct dilution method rapidly reduces the concentration of denaturant and protein by introducing a small volume of denatured protein solution into a large volume of refolding buffer. This instantaneous drop in denaturant concentration initiates refolding, while the simultaneous dilution of the protein minimizes intermolecular collisions that lead to aggregation [22] [35].
Detailed Protocol
- Solubilization: Isolate IBs by centrifugation and wash with a buffer containing Triton X-100 to remove membrane contaminants [35]. Solubilize the pellet in a solubilization buffer (e.g., 6–8 M GuHCl or urea, 50 mM Tris, 1 mM EDTA, pH 8.0) supplemented with a reducing agent (e.g., 10–100 mM DTT or 1% 2-mercaptoethanol) to break disulfide bonds. Incubate for 1–2 hours at room temperature with gentle agitation [40] [35].
- Clarification: Centrifuge the solubilized mixture at 15,000 × g for 20 minutes to remove any residual insoluble material.
- Refolding: Rapidly pipette the clarified supernatant into a vigorously stirred volume of pre-chilled refolding buffer (e.g., 50 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 8.0). A typical dilution factor is 100-fold, but this requires optimization [22] [35].
- Redox System: For disulfide-bonded proteins, include a redox system in the refolding buffer, such as reduced/oxidized glutathione (GSH/GSSG). A ratio of 2:1 to 10:1 (GSH:GSSG) is common, with total glutathione concentrations of 1–5 mM [36] [39].
- Incubation: Allow the refolding reaction to proceed for 12–48 hours at 4–10°C without agitation to minimize aggregation.
Workflow Diagram
Dialysis
Principle
Dialysis relies on the gradual removal of denaturants through diffusion across a semi-permeable membrane. The slow change in solvent composition allows the protein to explore its conformational landscape more gradually, potentially bypassing aggregation-prone states that occur at intermediate denaturant concentrations [22]. Step-wise dialysis, which uses a series of buffers with decreasing denaturant concentrations, offers even greater control [22].
Detailed Protocol
- Solubilization: Prepare the denatured protein as described in Section 3.1.2.
- Setup: Load the denatured protein solution into a dialysis membrane with an appropriate molecular weight cutoff.
- Refolding via Dialysis:
- One-Step Dialysis: Dialyze directly against a large volume (e.g., 1000x sample volume) of refolding buffer (e.g., 50 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 8.0). Change the buffer 2–3 times over 24–48 hours [22].
- Step-Wise Dialysis: For greater control, first dialyze against a buffer containing a medium concentration of denaturant (e.g., 2–4 M urea), then progressively step down to lower concentrations, and finally into a denaturant-free refolding buffer. Equilibrate for 4–8 hours at each step [22] [35].
- Redox Conditions: Include a redox pair like GSH/GSSG in the final dialysis buffer if the protein requires disulfide bond formation.
On-Column Refolding
Principle
On-column refolding, or matrix-assisted refolding, immobilizes the denatured protein on a solid support before initiating refolding. This physical separation of protein molecules prevents intermolecular aggregation during the critical refolding process [40] [36]. Immobilized Metal Ion Affinity Chromatography (IMAC) is commonly used for His-tagged proteins.
Detailed Protocol
- Solubilization and Binding: Solubilize IBs as in 3.1.2. Use a denaturing binding buffer (e.g., 6 M GuHCl, 20 mM imidazole, 0.5 M NaCl, 20 mM Tris, pH 8.0). Load the clarified supernatant onto an IMAC column (e.g., Ni-NTA) pre-equilibrated with the same buffer. The denatured protein binds to the resin via its affinity tag [36].
- Wash: Wash the column with 5–10 column volumes (CV) of binding buffer to remove unbound contaminants and residual reducing agents.
- Gradual Denaturant Removal: Apply a linear or step-wise gradient from the denaturing buffer to a native refolding buffer over 5–10 CV. The refolding buffer should lack denaturant and contain components that promote correct folding (e.g., arginine, glycerol, redox systems) [36].
- Final Wash and Elution: After the gradient, wash with an additional 5–10 CV of refolding buffer. Elute the refolded protein with an elution buffer containing high imidazole (e.g., 250–500 mM).
Workflow Diagram
The Scientist's Toolkit: Key Reagents and Materials
Successful refolding often depends on the use of specific additives that suppress aggregation, stabilize folding intermediates, or facilitate correct disulfide bond formation [22] [40] [36].
Table 2: Essential Research Reagents for Protein Refolding
| Reagent Category | Example Compounds | Primary Function & Mechanism |
|---|---|---|
| Chaotropic Denaturants | Urea, Guanidine HCl (GdnHCl) | Solubilize IBs by disrupting hydrophobic interactions; at low concentrations, can suppress aggregation [22] [40]. |
| Reducing Agents | Dithiothreitol (DTT), 2-Mercaptoethanol, TCEP | Reduce incorrect disulfide bonds in solubilized IBs to prepare for correct oxidative refolding [22] [35]. |
| Redox Pair Systems | Reduced/Oxidized Glutathione (GSH/GSSG), Cysteine/Cystine | Create a redox shuttle in the refolding buffer to catalyze the formation and isomerization of correct disulfide bonds [36] [39]. |
| Amino Acid Additives | L-Arginine, L-Proline | Suppress protein aggregation by weak association with folding intermediates; arginine is the most widely used [22] [40]. |
| Polyols and Sugars | Glycerol, Sucrose, Trehalose, PEG | Act as excluded solutes (kosmotropes) that stabilize the native protein state by preferential hydration [22] [40]. |
| Detergents & Surfactants | CHAPS, Triton X-100, N-Lauroylsarcosine | Solubilize IBs under mild conditions or shield hydrophobic patches on folding intermediates to prevent aggregation [40] [35]. |
| Artificial Chaperones | Cyclodextrins | First, a detergent (e.g., CTAB) binds to hydrophobic regions to prevent aggregation; then cyclodextrin strips away the detergent to allow controlled refolding [40]. |
Advanced Refolding and Analytical Considerations
Refolding with Chemical Additives and Artificial Chaperones
The strategic use of chemical additives is critical for improving refolding yields. Arginine, in particular, is highly effective at concentrations of 0.4–1.0 M in suppressing aggregation without inhibiting the folding reaction itself [22]. Artificial chaperone systems use a two-step process: first, a detergent like cetyltrimethylammonium bromide (CTAB) binds to the denatured protein to prevent aggregation; subsequently, a stripping agent like cyclodextrin is added to remove the detergent and allow the protein to refold in a controlled manner [40].
Quantitative Analytics for Refolding States
Monitoring refolding processes is challenging due to low protein concentrations and transient intermediates. A simplified kinetic model describes the dynamics between the folding intermediate (I), native (N), and aggregated (A) states [38]. Analytical techniques like HPLC and enzymatic activity assays are reference standards. High-throughput quantitative methods, such as MALDI-TOF-MS with 18O-labeled internal standards, can be used to optimize refolding conditions for disulfide-bonded proteins by accurately quantifying the formation of native disulfide bonds [39].
Dilution, dialysis, and on-column refolding represent three foundational methodologies for recovering active proteins from inclusion bodies. The choice of method depends on the protein's characteristics, available resources, and the required yield and concentration. The integration of chemical additives, particularly aggregation suppressors like arginine and optimized redox systems, is a powerful strategy to enhance the efficiency of any refolding process. As the field advances, the implementation of quantitative analytics and high-throughput screening will be essential for moving from empirical optimization to knowledge-driven, robust refolding platform strategies.
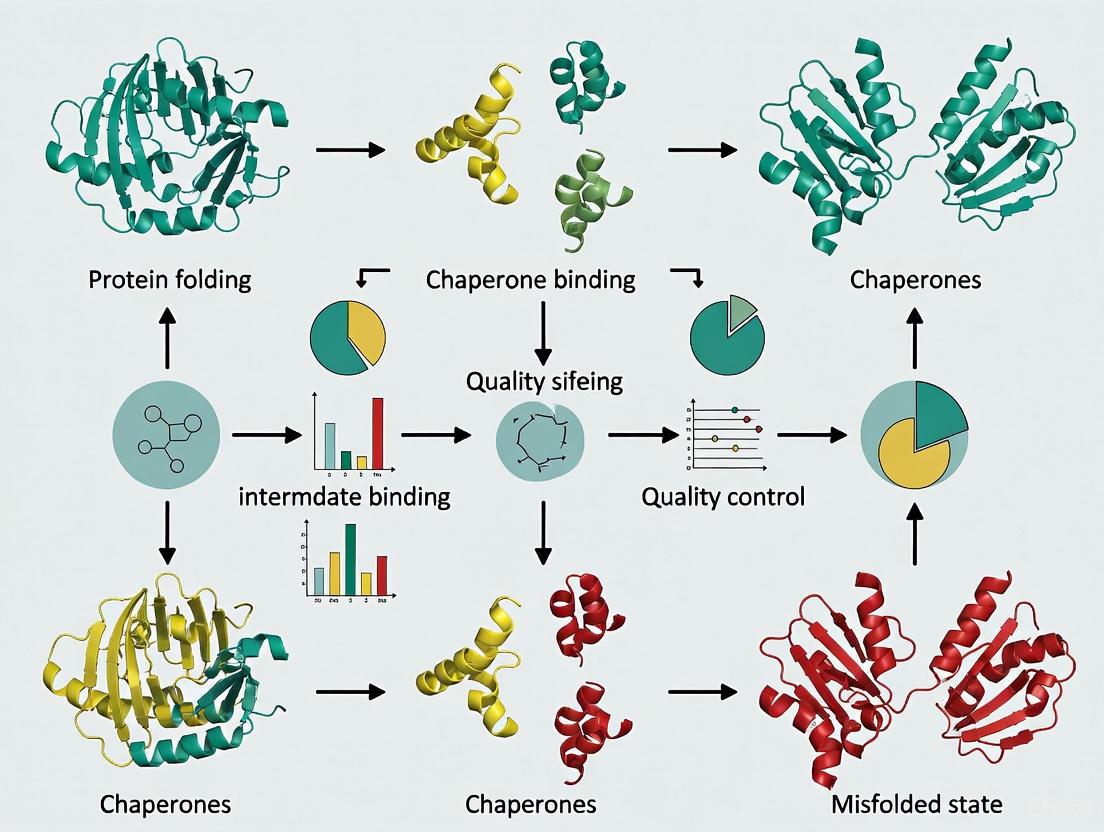
Within the broader research on chaperone-assisted protein refolding, establishing a baseline spontaneous refolding protocol is fundamental. This protocol details the methodology for recovering active Carbonic Anhydrase B (CAB) from its urea-denatured state without the assistance of folding modulators, providing a critical control for evaluating the efficacy of various chaperone systems [22]. The spontaneous refolding process is driven by the innate thermodynamic principle that a protein's native structure is the most stable under physiological conditions [3]. However, the journey from a denatured, unfolded chain to the correctly folded native state is often hampered by off-pathway events, particularly aggregation, which occurs when exposed hydrophobic surfaces interact irremediably [22] [41]. This protocol aims to minimize such aggregation through controlled denaturant removal, enabling researchers to quantify the baseline yield of active enzyme, against which the performance of artificial or natural chaperones can be measured [42].
Materials and Reagents
Research Reagent Solutions
The following table lists the essential reagents required for the denaturation and refolding of CAB.
Table 1: Key Research Reagents and Their Functions
| Reagent | Function in Protocol | Specification / Notes |
|---|---|---|
| Carbonic Anhydrase B (CAB) | Target protein for refolding studies | Isolated from bovine erythrocytes; >95% purity. |
| Urea | Chemical denaturant | Ultra-pure grade; fresh 8M solution prepared in Buffer A to avoid cyanate formation. |
| Dithiothreitol (DTT) | Reducing agent | Added to denaturation buffer to reduce and prevent incorrect disulfide bond formation. |
| Tris-HCl Buffer | Refolding buffer (Buffer A) | 50 mM, pH 8.5, serves as the physiological-like buffer for refolding [42]. |
| EDTA | Chelating agent | 1 mM in Buffer A; chelates metal ions that may catalyze oxidation. |
Methodology
Denaturation of Native CAB
- Preparation of Denaturation Solution: Prepare a denaturation buffer containing 8 M Urea, 50 mM Tris-HCl (pH 8.5), and 5 mM DTT.
- Denaturation Process: Add the native CAB protein to the denaturation buffer at a final concentration of 1 mg/mL.
- Incubation: Allow the mixture to incubate at room temperature (25°C) for 2 hours to ensure complete unfolding and reduction.
Spontaneous Refolding via Dilution
- Initiation of Refolding: To initiate refolding, rapidly dilute the denatured protein solution 100-fold into a large volume of pre-chilled Refolding Buffer (Buffer A: 50 mM Tris-HCl, pH 8.5, 1 mM EDTA).
- Final Conditions: This dilution reduces the urea concentration to a non-denaturing level (0.08 M) and the protein concentration to 10 μg/mL, a low concentration critical for minimizing intermolecular aggregation [22].
- Refolding Incubation: Allow the refolding reaction to proceed for 12-16 hours (overnight) at 4°C to maximize the recovery of active enzyme.
Activity Assay
- The enzymatic activity of refolded CAB is determined using a standard esterase activity assay with p-nitrophenyl acetate as the substrate.
- The refolding yield is calculated by comparing the activity of the refolded sample to an equivalent amount of native, undenatured CAB.
Expected Results and Data Analysis
Under the optimized conditions described above, the spontaneous refolding of CAB typically achieves a reactivation yield of <10% [42]. The majority of the protein forms aggregates or misfolded species that precipitate out of solution. The following table summarizes the key quantitative parameters and expected outcomes.
Table 2: Summary of Refolding Parameters and Expected Outcomes
| Parameter | Denatured State | Refolding Conditions |
|---|---|---|
| Urea Concentration | 8 M | ~0.08 M |
| Protein Concentration | 1 mg/mL | 10 μg/mL |
| Key Additives | 5 mM DTT | None (Spontaneous) |
| Incubation Temperature | 25°C | 4°C |
| Typical Incubation Time | 2 hours | 12-16 hours |
| Expected Yield of Active Protein | 0% | <10% |
The low yield underscores the challenges of spontaneous refolding, where the competition between correct folding and aggregation is often lost [22] [41]. This result highlights the necessity for assisted refolding strategies, such as the use of artificial chaperones, which can capture the unfolded protein and prevent aggregation, thereby increasing the reactivation yield to over 80% [42].
Workflow and Logical Diagram
The following diagram illustrates the critical steps and potential outcomes of the spontaneous refolding protocol, highlighting the decision points and the central role of aggregation.
Comparison with Artificial Chaperone-Assisted Refolding
The stark contrast in efficiency between spontaneous and assisted refolding justifies the research into chaperone systems. The following diagram and table detail the artificial chaperone process for direct comparison.
Table 3: Spontaneous vs. Artificial Chaperone-Assisted Refolding of CAB
| Characteristic | Spontaneous Refolding | Artificial Chaperone-Assisted Refolding |
|---|---|---|
| Principle | Thermodynamic driving force; trial-and-error folding [3]. | Sequential use of a detergent and cyclodextrin to capture and release the protein [42]. |
| Key Components | Refolding buffer only. | Cetyltrimethylammonium bromide (CTAB) and β-Cyclodextrin. |
| Mechanism | Direct dilution out of denaturant. | 1. CTAB captures unfolded protein, preventing aggregation. 2. Cyclodextrin strips detergent, inducing refolding [42]. |
| Typical Yield | <10% [42]. | >80% [42]. |
| Primary Advantage | Simple, requires minimal reagents. | High yield of active protein by effectively suppressing aggregation. |
| Primary Disadvantage | Very low yield; highly sensitive to protein concentration. | Requires optimization of detergent and cyclodextrin concentrations. |
Within the broader context of chaperone-assisted protein refolding research, the artificial chaperone protocol represents a bio-inspired in vitro strategy. This method mimics the two-step mechanism of natural chaperone systems like GroEL/GroES, utilizing readily available laboratory chemicals instead of complex protein machinery [43]. The core principle involves first using a detergent to capture and stabilize unfolded proteins, preventing irreversible aggregation. Subsequently, a cyclodextrin is introduced to strip away the detergent, creating a controlled environment that allows the protein to refold into its native, functional conformation [43] [44]. This protocol is particularly valuable for recovering active protein from aggregates or inclusion bodies, common challenges in recombinant protein production and in vitro studies [45].
Principle and Mechanism
Artificial chaperone-assisted refolding is designed to prevent aggregation by controlling the folding pathway. The following diagram illustrates the sequential, two-step mechanism of the process.
The mechanism relies on the specific properties of its components. The detergent acts as a "capture" agent, mimicking the function of holdase chaperones like Hsp70. Its amphiphilic nature allows it to interact with exposed hydrophobic patches on the unfolded protein, shielding these sticky surfaces and thereby preventing off-pathway interactions that lead to aggregation [43] [44]. In the second step, cyclodextrins act as "stripping" agents. Their hydrophobic interior cavity facilitates the formation of inclusion complexes with the detergent molecules, efficiently removing the detergent coat from the protein surface [46]. This controlled, gradual removal creates a permissive environment for the protein to explore its conformational landscape and attain its native fold [43].
Reagents and Equipment
The Scientist's Toolkit: Essential Research Reagents
The following table details the key reagents required to execute the artificial chaperone refolding protocol.
Table 1: Key Research Reagent Solutions for Artificial Chaperone Refolding
| Reagent Category | Specific Examples | Function & Mechanism |
|---|---|---|
| Denaturant | Guanidine HCl (GdmCl), Urea | Unfolds the target protein from a solid aggregate (e.g., inclusion bodies) to create a starting solution of denatured polypeptide. |
| Detergent (Capture Agent) | Cetyltrimethylammonium bromide (CTAB), Sodium Dodecyl Sulfate (SDS) | Binds to hydrophobic regions of the unfolded protein, preventing aggregation by shielding interactive surfaces [43]. |
| Cyclodextrin (Stripping Agent) | α-Cyclodextrin, β-Cyclodextrin | Strips the detergent from the protein by forming an inclusion complex with it, enabling controlled refolding [43] [44]. |
| Refolding Buffer | Tris-HCl, Phosphate Buffered Saline (PBS) | Provides the optimal pH, ionic strength, and redox conditions (if disulfide bonds are present) for the target protein to attain its native conformation. |
Required Equipment
- Standard Biophysics Lab Equipment: UV-Visible spectrophotometer with temperature control (for turbidity and activity assays), circular dichroism (CD) spectrometer, fluorimeter.
- Chromatography Systems: Fast Protein Liquid Chromatography (FPLC) or HPLC system for purification and analysis.
- Standard Labware: Temperature-controlled water bath or incubator, vortex mixer, precision pipettes, and a variety of tubes and containers.
Step-by-Step Experimental Protocol
Sample Preparation and Denaturation
- Solubilize Inclusion Bodies: Isolate inclusion bodies from recombinant E. coli culture via centrifugation. Wash thoroughly with a buffer containing 2 M urea and 1-2% Triton X-100 to remove membrane components. Centrifuge again and discard the supernatant.
- Denature Protein: Solubilize the pellet in a denaturation buffer (e.g., 50 mM Tris-HCl, pH 8.5, 6 M GdmCl, 10 mM DTT). Incubate for 1-2 hours at room temperature with gentle agitation.
- Clarify Solution: Centrifuge the denatured protein solution at 15,000 × g for 20 minutes to remove any insoluble debris. Transfer the supernatant, which contains the denatured protein, to a fresh tube.
Artificial Chaperone Refolding
The core refolding procedure is a sequential two-step process. The table below provides a generalized protocol, with specific examples from the literature.
Table 2: Artificial Chaperone Refolding Procedure and Conditions
| Step | Action | Parameters & Examples from Literature |
|---|---|---|
| 1. Capture | Add detergent to the denatured protein solution. Mix gently but thoroughly. | Typical Conditions: Incubate at a slightly elevated temperature (e.g., 50°C for 15 min) or at room temperature. Example: For carbonic anhydrase B, citrate synthase, and lysozyme, incubation was performed with the detergent under conditions that would normally lead to aggregation [43]. |
| 2. Refolding | Initiate refolding by adding a concentrated stock solution of cyclodextrin to the protein-detergent mixture. | Typical Conditions: Refolding proceeds for several hours at room temperature or 4°C. Example: Cyclodextrin is added to trigger the removal of detergent from the protein-detergent complex, allowing the protein to refold [43]. Specific Case: For SDS-induced amyloid fibrils of myoglobin, the addition of β-cyclodextrin in a 20:1 molar ratio (CD:SDS) significantly reduced turbidity and promoted refolding to a native-like state [44]. |
Post-Refolding Purification and Analysis
After refolding is complete, the protein solution must be processed to remove the artificial chaperone components and isolate the correctly folded protein.
- Remove Aggregates: Pass the refolding mixture through a 0.22 µm filter to remove any large aggregates that may have formed [43].
- Separate from Chaperones: Perform buffer exchange or diafiltration using a centrifugal filter unit with an appropriate molecular weight cutoff (MWCO). A MWCO of 10 kDa is effective for retaining the refolded enzyme while allowing low-molecular-weight artificial chaperones (detergent and cyclodextrin) to pass through [43].
- Concentrate and Store: Concentrate the purified, refolded protein using a centrifugal filter and store it in an appropriate storage buffer at recommended temperatures.
Expected Results and Data Interpretation
Successful refolding is characterized by a decrease in solution turbidity, recovery of enzymatic activity, and spectroscopic signatures of the native structure.
Table 3: Key Assays for Monitoring Refolding Success
| Assay | What to Measure | Interpretation of Results |
|---|---|---|
| Turbidity | Absorbance at 400 nm (A₄₀₀) or 360 nm (A₃₆₀). | A high value indicates light scattering due to large aggregates. Success is marked by a significant decrease in turbidity after the cyclodextrin step [44]. |
| Activity Assay | Enzyme-specific catalytic activity. | The recovery of enzymatic function is the ultimate indicator of success. Compare activity to a native standard. |
| Circular Dichroism (CD) | Far-UV CD spectrum (190-250 nm). | A spectrum that matches the known native structure (e.g., characteristic minima for α-helices or β-sheets) indicates proper secondary structure refolding [44]. |
| Fluorescence Spectroscopy | Intrinsic Tryptophan fluorescence. | A shift in the emission maximum and/or change in intensity towards that of the native protein indicates a proper tertiary packing environment. |
Troubleshooting and Optimization
The following flowchart provides a systematic approach to diagnosing and resolving common issues encountered during the refolding process.
Key optimization parameters include:
- Detergent and Cyclodextrin Selection: The choice and ratio of detergent to cyclodextrin are critical. While CTAB/α-cyclodextrin is a common pair, SDS/β-cyclodextrin has been effective for dissolving amyloid fibrils [44]. The optimal ratio must be determined empirically.
- Protein Concentration: Refolding yields are highly dependent on protein concentration. Start with low concentrations (0.01-0.1 mg/mL) to minimize aggregation and scale up optimistically.
- Buffer Conditions: The pH, ionic strength, and composition of the refolding buffer can significantly impact the efficiency of folding. The inclusion of redox agents like glutathione is essential for proteins requiring disulfide bond formation.
The folding of proteins into their native, functional three-dimensional structures is a fundamental process in biology. While many small, single-domain proteins can fold spontaneously, the crowded cellular environment and the inherent complexity of larger proteins often necessitate assistance from molecular chaperones [41]. Among these, the GroEL/GroES chaperonin system is essential, providing a central folding chamber for a diverse range of cellular proteins [41]. Studying chaperone-assisted folding in vitro is crucial for deciphering these complex pathways. This protocol details the use of a slow-folding GFP mutant (sGFP), an ideal model substrate whose fluorescence is directly tied to its correctly folded state, enabling real-time monitoring of GroEL/ES-assisted refolding [47]. This method provides researchers with a robust system to investigate the effects of various conditions and co-factors on the protein folding pathway.
Theoretical Background
The Protein Folding Challenge and Molecular Chaperones
Protein folding in vivo is challenged by molecular crowding, which increases the risk of off-pathway events like aggregation and misfolding [41]. The cellular proteostasis network, which includes molecular chaperones, has evolved to mitigate these risks. Chaperones are categorized as ATP-independent "holdases," which prevent aggregation, and ATP-dependent "foldases," which actively promote folding [41]. The GroEL/GroES system is a key ATP-dependent chaperonin that provides a central folding chamber for many cellular proteins [41].
The GroEL/GS Chaperonin System
The GroEL/GroES system functions as a molecular machine that undergoes ATP-driven conformational changes to facilitate folding. GroEL is a large double-ring tetradecamer, with each subunit containing an ATPase domain. GroES is a single-ring heptamer that acts as a lid for the GroEL cavity. The current model suggests that GroEL does not necessarily accelerate the folding process itself, but rather prevents irreversible aggregation by binding unfolded protein substrates and providing them with a protected, isolated environment to undergo spontaneous folding [4]. This encapsulated chamber helps proteins navigate their folding energy landscape by preventing inappropriate interactions.
sGFP as a Folding Reporter
Wild-type Green Fluorescent Protein (GFP) and its variants fold on a time scale of minutes, with the slow step often involving cis-trans peptide bond isomerization, particularly at conserved proline residues [48]. The sGFP used in this protocol is a slow-folding mutant whose fluorescence is directly correlated with its native structure. It is fluorescent only when correctly folded, providing a direct, real-time readout of the refolding process without relying on enzymatic activity or intrinsic tryptophan fluorescence [47]. This makes it a superior tool for kinetic studies of protein folding.
Materials and Reagents
Research Reagent Solutions
The following table lists the essential materials and reagents required to perform the spontaneous and chaperonin-assisted refolding of sGFP.
Table 1: Essential Reagents and Materials for sGFP Refolding Assay
| Item | Function/Brief Explanation |
|---|---|
| Purified sGFP protein | The slow-folding mutant substrate protein for refolding studies [47]. |
| Guanine Hydrochloride (GuHCl) | A chemical denaturant used to completely unfold the native sGFP protein [47]. |
| Buffer A | The refolding buffer; provides a folding-favorable environment upon dilution of the denaturant [47]. |
| GroEL tetradecamers | The core component of the chaperonin system; forms the central folding cage [47]. |
| GroES heptamers | The co-chaperonin; acts as a lid for the GroEL cavity, encapsulating the substrate [47]. |
| ATP | The energy source required to drive the conformational changes in the GroEL/GroES folding cycle [47]. |
| Fluorescence Spectrophotometer | Instrument used to monitor the increase in fluorescence at 515 nm over time [47]. |
| Quartz Cuvette | Housing for the refolding reaction during fluorescence measurement [47]. |
Equipment
- Pipettes (2.5-μl, 10-μl, 200-μl, 1-ml)
- Centrifuge
- Fluorescence spectrophotometer (e.g., Fluorolog 3 Spectrofluorometer, Jobin Yvon, Horiba) [47]
- Dry bath
- pH meter
- Microcentrifuge tubes (1.5-ml), Falcon tubes (15-ml), and a fluorescence cuvette (3.0 mm path length recommended) [47]
Recipes
- Buffer A: Specific composition as used in the cited protocol. (Note: The exact concentrations of Tris, KCl, MgCl₂, DTT, etc., should be sourced directly from the original protocol [47]).
Experimental Protocol
The diagram below illustrates the key steps involved in the GroEL/ES-assisted refolding of sGFP, from unfolding to fluorescence measurement.
Detailed Step-by-Step Procedure
A. Protein Unfolding
- In a 1.5 ml microcentrifuge tube, add 2.5 μl of sGFP (20 μM) to 7.5 μl of 8 M GuHCl in Buffer A. The final concentration of GuHCl in the unfolding reaction will be 6 M [47].
- Incubate the tube in a dry bath at 25°C for 1 hour to achieve complete unfolding of the sGFP protein [47].
B. Spontaneous Refolding (Control Experiment)
- Instrument Setup: Switch on the fluorescence spectrophotometer and allow the lamp to warm up for 45 minutes to minimize fluctuations. Set the instrument to kinetic acquisition mode with the following parameters [47]:
- Excitation wavelength: 480 nm
- Emission wavelength: 515 nm
- Collection interval: 20 s
- Integration time: 0.7 s
- Total kinetic time: 1800 s
- Temperature: 25°C
- Refolding Reaction: Clean a quartz cuvette with methanol and MilliQ water. Dilute 2 μl of the unfolded sGFP protein 100-fold into 198 μl of Buffer A (final volume 200 μl) directly in the cuvette. Mix thoroughly by pipetting [47].
- Data Acquisition: Start the kinetics acquisition immediately after mixing.
C. Chaperonin-Assisted Refolding
- Instrument Setup: Follow the same instrument setup procedure as for spontaneous refolding (Step B1) [47].
- Refolding Reaction: In a clean quartz cuvette, combine the following components to a final volume of 200 μl [47]:
- 2 μl (200 nM final) of completely unfolded sGFP.
- Buffer A containing GroEL (400 nM tetradecamers).
- GroES (800 nM heptamers).
- ATP (2 mM final concentration) to initiate refolding.
- Note: The recommended molar ratio of substrate:GroEL:GroES is 1:2:4 [47].
- Mix the reaction thoroughly by slow pipetting and begin kinetics acquisition immediately.
Data Analysis and Interpretation
Kinetic Model and Fitting
The refolding kinetics of sGFP typically follows a two-state pathway. The fluorescence data obtained should be fitted using nonlinear regression with a single exponential equation [47]:
y = y₀ + A₁ * e^(-x/t₁)
Where:
- y is the fluorescence intensity at time x.
- y₀ is the offset value.
- A₁ is the amplitude.
- t₁ is the time constant.
The apparent refolding rate (k), is calculated as k = 1 / t₁ [47].
Expected Results and Quantification
The table below summarizes the key parameters expected from spontaneous versus chaperonin-assisted refolding experiments, allowing for direct comparison.
Table 2: Key Quantitative Parameters from Refolding Experiments
| Parameter | Spontaneous Refolding | GroEL/ES-Assisted Refolding | Explanation & Significance |
|---|---|---|---|
| Apparent Refolding Rate (k) | Measured value (min⁻¹) | Measured value (min⁻¹) | The reciprocal of the time constant (1/t₁); indicates the speed of the folding reaction [47]. |
| Refolding Amplitude | Measured value (A.U.) | Measured value (A.U.) | The total increase in fluorescence; correlates with the final yield of correctly folded protein [47]. |
| Kinetically Trapped State | May be present [48] | Often minimized | Non-fluorescent, misfolded state; GroEL/ES can help proteins avoid this trap [48]. |
The GroEL/ES Functional Cycle
The following diagram illustrates the functional cycle of the GroEL/GroES chaperonin system during substrate protein folding, based on the mechanistic insights provided in the search results [41] [4].
Troubleshooting
- Low Fluorescence Signal: Ensure the protein was completely unfolded. Verify the purity and concentration of GroEL, GroES, and ATP. Check instrument settings and lamp functionality [47].
- High Background Noise: Allow the fluorometer lamp to warm up sufficiently (45 min). Use high-quality, clean cuvettes [47].
- No Refolding Observed: Confirm the activity of the chaperonin proteins and the freshness of the ATP solution. Ensure the dilution factor and reagent concentrations are accurate [47].
Within the framework of chaperone-assisted protein refolding research, maintaining proteostasis—the delicate balance between protein synthesis, folding, and degradation—is paramount for cellular health and function [29]. Disruptions to this equilibrium, termed dysproteostasis, lead to protein misfolding and aggregation, underlying numerous human diseases from neurodegenerative disorders to cancer [29]. While molecular chaperones like HSP70 and TRiC are specialized proteins that assist folding in vivo, chemical chaperones represent a class of small molecules that stabilize protein conformations and suppress aggregation in diverse experimental and therapeutic contexts [49] [50]. These compounds function through nonspecific mechanisms, primarily by promoting favorable protein-water interactions and stabilizing native protein structures against denaturing stresses [50]. This application note provides a structured overview of commonly employed chemical chaperones, quantitative data on their efficacy, and detailed protocols for their implementation in refolding experiments, specifically tailored for researchers and drug development professionals.
Theoretical Foundations: Mechanisms of Action
Classification and General Properties
Chemical chaperones are broadly categorized as compatible osmolytes or pharmacological chaperones, with this document focusing on the former. Compatible osmolytes are naturally occurring small organic molecules accumulated by cells under stress conditions to protect macromolecular structure and function [49]. They are termed "compatible" because they do not perturb cellular components at high concentrations. Their primary mechanism involves preferential exclusion from the protein's surface, which thermodynamically favors the more compact, native state over unfolded or misfolded states that have greater surface area [49]. This creates a free energy bias toward the properly folded conformation, effectively increasing the kinetic barrier to aggregation.
Contrasting Molecular and Chemical Chaperones
While both molecular and chemical chaperones mitigate aggregation, their modes of action differ significantly, as summarized in the table below.
Table 1: Comparison of Molecular versus Chemical Chaperones
| Feature | Molecular Chaperones (e.g., GroEL, DnaK) | Chemical Chaperones (Osmolytes) |
|---|---|---|
| Chemical Nature | Proteins (often ATP-dependent) [29] [51] | Small molecules (e.g., polyols, amino acids) [49] [50] |
| Specificity | Often specific to client proteins or pathways [51] [52] | General, non-specific stabilization [50] |
| Primary Mechanism | Active, ATP-driven binding and release cycles [53] | Preferential exclusion & thermodynamic stabilization [49] |
| Typical Use Context | In vivo folding, co-translational folding [29] [54] | In vitro assays, storage buffers, therapeutic formulations [50] [55] |
Figure 1: Chaperone Mechanisms in Protein Folding. Chemical chaperones act indirectly by altering the solvent environment to favor the native state, while molecular chaperones directly bind to client proteins to prevent aggregation and facilitate folding.
Quantitative Data on Common Chemical Chaperones
The efficacy of a chemical chaperone depends on the specific protein system and the environmental conditions. The following table synthesizes data on commonly used compounds, their stabilization mechanisms, and typical application contexts.
Table 2: Profile of Common Chemical Chaperones and Their Applications
| Chaperone | Class | Reported Mechanism | Effective Concentration Range | Typical Use Cases |
|---|---|---|---|---|
| Glycerol [50] | Polyol | Preferential hydration; reduces molecular mobility | 5-20% (v/v) | Enzyme storage buffers; PCR mixes; prevents freeze-thaw damage |
| Trehalose [49] [50] | Sugar | Forms glassy matrix; replaces water H-bonds | 0.1-0.5 M | Lyophilization stabilizer; long-term protein storage |
| Proline [49] | Amino Acid | Preferentially excluded; suppresses aggregation | 0.1-1.0 M | In vitro refolding of inclusion bodies |
| Betaine [49] [50] | Methylamine | Osmotic regulator; stabilizes folded proteins | 0.1-0.5 M | Stress protection in cell culture; protein crystallization |
| Arginine | Amino Acid | Suppresses aggregation; not typically stabilizing | 0.1-0.5 M | Suppresses aggregation during refolding; elution buffer in chromatography |
| BSA [50] | Protein | Nonspecific adsorption of aggregates | 0.1-1.0 mg/mL | Restriction enzyme digests; antibody assays |
Practical Applications and Protocols
Aggregation Prevention Assay
The aggregation prevention assay is a definitive method to determine chaperone activity by monitoring the suppression of heat-induced protein aggregation via light scattering [53].
Materials and Reagents:
- Test Protein Substrate: Citrate Synthase (CS) from porcine heart or Maltodextrin glucosidase (MalZ) [53].
- Positive Control Chaperone: Purified GroEL (0.5 mg/mL) [53].
- Negative Control: Lysozyme (1 mg/mL), which lacks general chaperone activity [53].
- Chemical Chaperones: Glycerol, Trehalose, Arginine-HCl, etc.
- Assay Buffer: 50 mM Tris-HCl, pH 8.0.
- Equipment: Fluorescence spectrophotometer with a temperature-controlled Peltier cuvette holder and a stirred quartz cuvette.
Procedure:
- Sample Preparation: In a microcentrifuge tube, prepare a 0.75 mL reaction in assay buffer containing:
- 0.15 µM Citrate Synthase (CS) [53].
- The chemical chaperone at the desired concentration (e.g., 0.4 M Arginine, 10% Glycerol). Note: The optimal chaperone-to-substrate ratio must be determined empirically.
- Instrument Setup: Pre-equilibrate the spectrophotometer and the cuvette chamber to 45-47°C. Set the excitation and emission wavelengths to 320 nm (light scattering mode) with slits at 2-5 nm.
- Data Acquisition: Transfer the mixture to a pre-warmed quartz cuvette. Start the time course measurement, taking a light scattering reading every 1-2 minutes for 50-60 minutes. Gently mix the solution by see-saw movement between readings if a magnetic stirrer is unavailable.
- Analysis: Plot the light scattering intensity (arbitrary units) versus time. Compare the initial slopes and final plateaus of the curves with the no-chaperone control. Effective chaperones will show a significant reduction in light scattering signal.
Suppression of Aggregation During Refolding
This protocol uses chemical chaperones to increase the yield of properly folded protein during the dilution-refolding of denatured proteins, such as from inclusion bodies.
Materials and Reagents:
- Denatured Protein: Target protein denatured in 6 M Guanidine HCl or 8 M Urea.
- Refolding Buffer: 50 mM Tris, 100 mM NaCl, 2 mM EDTA, 1 mM GSH, 0.5 mM GSSG, pH 8.0.
- Chemical Chaperones: Prepare stock solutions of Arginine-HCl (2 M, pH 7.5), Glycerol (80% v/v), and Trehalose (1 M).
- Dialysis Tubing or Desalting Columns.
Procedure:
- Refolding Initiation: Rapidly dilute the denatured protein stock 50- to 100-fold into pre-chilled refolding buffer. Immediately supplement the refolding buffer with chemical chaperones. Suggested final concentrations: 0.4-0.8 M Arginine, 10% Glycerol, or 0.2 M Trehalose.
- Incubation: Allow refolding to proceed for 4-16 hours at 4°C.
- Chaperone Removal: After refolding, remove the chemical chaperones via dialysis against a standard storage buffer or using desalting size-exclusion chromatography. This step is crucial as some osmolytes like arginine can inhibit the function of the final folded protein.
- Analysis: Assess refolding yield and aggregation suppression by:
- Size-Exclusion Chromatography (SEC): To quantify monomeric protein and soluble aggregates.
- Dynamic Light Scattering (DLS): To monitor the hydrodynamic radius of the protein population.
- Activity Assay: To confirm functional recovery.
Figure 2: Experimental Workflow for Refolding with Chemical Chaperones. Incorporating chemical chaperones into the refolding buffer biases the pathway toward productive folding and away from off-pathway aggregation.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and materials essential for conducting experiments on chemical chaperone-assisted protein refolding.
Table 3: Essential Research Reagents for Chaperone Studies
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| Model Aggregation-Prone Substrates | Validating chaperone activity in vitro. | Citrate Synthase (CS) [53], Maltodextrin glucosidase (MalZ) [53]. |
| Molecular Chaperone Standards | Positive controls for activity assays. | GroEL/ES (Hsp60) [51] [53], DnaK/DnaJ/GrpE (Hsp70/40) [51]. |
| Common Chemical Chaperones | Nonspecific stabilization in buffers. | Glycerol, Trehalose, L-Arginine-HCl, Betaine, Proline [49] [50]. |
| Denaturants | Unfolding proteins for refolding studies. | Urea, Guanidine Hydrochloride (GuHCl) [49]. Note: GuHCl is a denaturing osmolyte. |
| Spectrophotometer with Peltier | Real-time monitoring of aggregation. | Equipped with a stirred cuvette for light scattering assays [53]. |
| Chromatography Systems | Analyzing refolding success and purity. | Size-Exclusion Chromatography (SEC) for separating monomers from aggregates [55]. |
Chemical chaperones such as osmolytes and arginine are indispensable tools in the researcher's arsenal for combating protein aggregation. Their ability to modulate the folding landscape through nonspecific thermodynamic mechanisms provides a powerful, versatile strategy for improving protein stability and refolding yields in vitro. The protocols and data summarized herein offer a practical foundation for integrating these compounds into refolding workflows, thereby enhancing the robustness and reproducibility of protein-based research and therapeutic development.
Solving Refolding Challenges: Aggregation, Low Yield, and Optimization
Identifying and Overcoming Kinetic Traps and Irreversible Aggregation
In the field of protein science, kinetic traps are metastable, non-native conformational states that can arrest the folding process, leading to irreversible aggregation and a significant loss of functional protein yield. This is a major challenge in both basic research and industrial applications involving protein refolding. Kinetic traps arise from the rugged nature of the protein folding energy landscape, where partially folded intermediates can become stranded in local energy minima, unable to overcome the activation barrier required to reach the native state. The subsequent accumulation and self-association of these misfolded species often result in amorphous aggregates or highly ordered amyloid fibrils, which are biologically inactive and can exhibit cytotoxic properties. Within the context of chaperone-assisted refolding protocols, understanding the molecular origin of these traps and developing robust methods to circumvent them is paramount for optimizing the recovery of functional proteins. This application note provides a detailed experimental framework for identifying kinetic traps and outlines proven chaperone-based strategies to prevent irreversible aggregation.
Theoretical Background: The Energy Landscape of Protein Folding
The concept of a folding funnel provides a useful theoretical model for visualizing how proteins navigate the journey from an unfolded chain to a uniquely folded native structure. In this model, the width represents the conformational entropy (the number of possible conformations), and the depth represents the energy, with the native state at the global minimum. A perfectly smooth funnel would allow a protein to fold rapidly and efficiently. However, in reality, the landscape is often rugged, featuring bumps, valleys, and local minima [29]. These local minima represent kinetic traps– partially folded states that are stable enough to persist but are not the functional native state.
The formation of a kinetic trap is characterized by a small energy difference (ΔG₁) between the correctly folding pathway and the off-pathway trap during the initial stages. However, once the protein enters the trapped state, a much larger energy barrier (ΔG₂) must be overcome to escape and continue productive folding [56]. This relationship is summarized in the table below.
Table 1: Energetic Parameters of a Kinetic Trap in S-layer Protein Assembly
| Energetic Parameter | Description | Experimental Value |
|---|---|---|
| ΔG₁ | Difference in formation energy barrier between the trapped state and the native state. | ~0.7 kT (approx. 1.6 kJ/mol) [56] |
| ΔG₂ | Activation energy barrier for the transformation from the trapped state to the native state. | ~25 kT (approx. 61 kJ/mol) [56] |
This small difference in initial formation energy means that the system has nearly equal probability of entering the productive folding pathway or falling into the kinetic trap. The极高的energy barrier for escape, however, renders the trapped state long-lived and prone to aggregation over time. The following diagram illustrates this landscape and the role of chaperones.
Diagram 1: Protein folding energy landscape with a kinetic trap. The dashed line indicates chaperone action that facilitates escape.
Experimental Identification and Quantification of Kinetic Traps
Key Experimental Signatures
Kinetic traps manifest through specific, measurable deviations from ideal two-state folding kinetics. The primary experimental signatures include:
- Non-Linear Chevron Plot Rollover: The chevron plot displays the logarithm of the observed folding/unfolding rate constant (lnkₒbₛ) against denaturant concentration. In two-state folding, the arms of the "V" are linear. A downward curvature or "rollover" in the folding limb (at low denaturant) is a classic indicator of a kinetic trap, suggesting the presence of a stable intermediate or a change in the rate-limiting step [57].
- Slow Phases in Refolding Kinetics: The appearance of multiple, slow kinetic phases during refolding, monitored by techniques like stopped-flow fluorescence, can indicate that a population of molecules is slowly converting from a trapped state to the native state.
- Direct Observation of Metastable States: Techniques with single-molecule resolution, such as in situ Atomic Force Microscopy (AFM), can directly visualize and distinguish between transient, metastable states and the final stable state, as demonstrated in S-layer protein assembly [56].
Protocol: Stopped-Flow Kinetics to Detect Rollover
Objective: To record a chevron plot and identify the presence of kinetic traps via rollover in the folding limb.
Materials:
- Purified protein of interest (>95% purity).
- Urea or Guanidine Hydrochloride (GdnHCl) stock solutions (ultrapure).
- Stopped-flow instrument equipped with fluorescence detection.
- Buffer components (e.g., 50 mM HEPES or Phosphate, pH 7.0).
Method:
- Sample Preparation:
- Prepare a concentrated stock of unfolded protein in a high concentration of denaturant (e.g., 8 M GdnHCl).
- Prepare a series of refolding buffers with varying, low concentrations of denaturant (e.g., 0.1 M to 2.0 M GdnHCl), all in standard consensus buffer (50 mM buffer, pH 7.0, 25°C) [57].
- Data Collection:
- Load the unfolded protein solution and a refolding buffer into the stopped-flow syringes.
- Rapidly mix the solutions (typical 1:10 ratio) and monitor the change in intrinsic tryptophan fluorescence or another suitable signal over time.
- Repeat this process for each denaturant concentration in the refolding series.
- Data Analysis:
- Fit the resulting kinetic traces to single or multi-exponential functions to extract the observed rate constant (kₒbₛ) for each condition.
- Plot lnkₒbₛ against the final denaturant concentration after mixing. Fit the unfolding limb with a linear regression. Visually inspect the folding limb for downward curvature (rollover).
- For quantitative analysis, fit the entire chevron to a model that accounts for rollover, such as a polynomial function or a specific kinetic mechanism [57].
Overcoming Kinetic Traps with Chaperone-Assisted Refolding
Molecular chaperones are the cell's natural solution to the problems of misfolding and aggregation. They do not provide steric information for folding but instead prevent off-pathway interactions and, in some cases, actively remodel kinetically trapped states.
Mechanism of Chaperone Action
Chaperones employ diverse mechanisms, which can be categorized as follows:
- Holdases: Bind to hydrophobic patches on non-native proteins (e.g., DnaJ/Hsp40), preventing aggregation by shielding them from the crowded cellular environment [58] [59].
- Foldases: Provide a secluded environment for folding. The GroEL/GroES (Hsp60/chaperonin) system is a prime example, encapsulating a single polypeptide chain in an ATP-dependent manner, allowing it to fold in isolation without the risk of aggregation [58] [59].
- Disaggregases: Machines like Hsp104/ClpB can disentangle already formed aggregates, returning proteins to a soluble state for another chance to fold [59].
A key insight from recent studies is that efficient folding while chaperone-bound requires weak chaperone-client interactions. Strengthening this interaction can inhibit the conformational mobility needed for the client protein to fold, turning the chaperone into a holdase rather than a foldase [60].
Protocol: GroEL/GroES (Hsp60/Hsp10) Assisted Refolding
Objective: To refold an aggregation-prone protein using the chaperonin system to bypass kinetic traps.
Materials:
- E. coli GroEL and GroES proteins (commercially available or purified).
- Aggregation-prone target protein, denatured in 6 M GdnHCl.
- Refolding buffer: 50 mM HEPES-KOH, pH 7.5, 50 mM KCl, 20 mM MgCl₂.
- ATP regeneration system: 2 mM ATP, 20 mM Creatine Phosphate, 50 µg/mL Creatine Kinase.
Method:
- Form the GroEL–Unfolded Protein Complex:
- Dilute the denatured target protein rapidly into a molar excess of GroEL (typically 1:1 to 1:2 complex ratio) in refolding buffer on ice. Incubate for 10-15 minutes to allow binding.
- Initiate Refolding:
- To the GroEL–protein complex, add a 2-5 fold molar excess of GroES (over GroEL rings) and the ATP regeneration system.
- Transfer the reaction to 25°C to initiate folding.
- Monitor Folding and Release:
- Monitor the recovery of native protein over time (minutes to hours) using enzymatic activity assays or native PAGE.
- Control reactions should include:
- No chaperones.
- GroEL without GroES or ATP.
- GroEL/GroES with a non-hydrolyzable ATP analog (e.g., ATPγS) to confirm ATP-dependence.
- Analysis:
- Compare the final yield of active protein and the kinetics of its appearance between the chaperone-assisted and control refolding reactions.
Table 2: Research Reagent Solutions for Chaperone-Assisted Refolding
| Reagent / Material | Function / Role in Protocol | Key Characteristics & Considerations |
|---|---|---|
| GroEL (Hsp60) | Core chaperonin; forms a double-ring structure that binds unfolded polypeptides. | Essential to have a functional, ATPase-active preparation. Purity >95%. |
| GroES (Hsp10) | Co-chaperonin; acts as a "lid" for GroEL, creating an encapsulated folding chamber. | Must be in a functional heptameric state. |
| ATP Regeneration System | Maintains a constant, high level of ATP for multiple rounds of GroEL action. | Prevents depletion of ATP, which would stall the refolding cycle. |
| Urea / GdnHCl | Chemical denaturant used to prepare the unfolded starting material. | Ultrapure grade to avoid chemical modifications. Concentration must be accurately determined. |
| Stopped-Flow Apparatus | For rapid mixing and monitoring of fast folding kinetics (milliseconds to seconds). | Requires a sensitive fluorescence detector. |
Advanced Technique: HDX-MS for Mapping Cotranslational Folding Intermediates
Kinetic traps can also form during protein synthesis on the ribosome. Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) is a powerful technique for resolving the structure and dynamics of nascent polypeptide chains associated with the ribosome and chaperones.
The following diagram outlines the key steps in an HDX-MS experiment to study chaperone-assisted folding on the ribosome.
Diagram 2: HDX-MS workflow for analyzing protein folding intermediates.
Protocol Highlights [25]:
- Sample Preparation: Generate stable, homogeneous RNCs with the nascent chain (NC) stalled at specific lengths. Purify complexes to remove free chaperones and other contaminants.
- Deuteration: Initiate exchange by diluting the RNCs into deuterated buffer for a defined time (e.g., 10 s, 100 s, 1000 s). This labels unstructured and dynamic regions of the NC.
- Quenching: Rapidly lower the pH to ~2.5 and the temperature to 0°C to minimize back-exchange.
- Digestion and Analysis: Digest the entire mixture (ribosomal proteins, NC, chaperones) with an acid-tolerant protease (e.g., pepsin). Separate the resulting peptides by liquid chromatography and analyze their mass increase due to deuterium incorporation.
- Data Interpretation: Peptides derived from structured, protected regions of the NC will show low deuterium uptake. Peptides from unstructured or dynamically exposed regions will show high uptake. Comparing the HDX patterns of RNCs of different lengths, with and without chaperones like Trigger Factor, reveals the folding pathway and how chaperones influence it by stabilizing or destabilizing specific intermediates [25].
Kinetic traps present a significant barrier to efficient protein folding in vitro and in vivo. Their identification relies on careful kinetic analysis, such as the observation of chevron plot rollover. The strategic use of molecular chaperones, such as the GroEL/GroES system, provides a powerful methodological solution by preventing aggregation and offering a controlled environment for proteins to navigate their energy landscape. Furthermore, advanced techniques like HDX-MS offer unprecedented peptide-level insight into how chaperones interact with nascent chains on the ribosome to guide folding and avoid kinetic traps. The protocols outlined herein provide a foundation for researchers to diagnose and overcome these challenges, thereby enhancing the efficacy of protein refolding strategies in both basic research and biopharmaceutical development.
Within the framework of chaperone-assisted protein refolding research, the precise control of biochemical parameters is not merely beneficial but fundamental to success. The process of converting an unfolded or misfolded polypeptide into its native, biologically active conformation is a delicate undertaking, perpetually in competition with off-pathway reactions that lead to irreversible aggregation and yield loss [35]. This document provides detailed application notes and protocols, grounded in current structural and mechanistic studies, to guide researchers in the systematic optimization of pH, temperature, ionic strength, and redox conditions. These parameters collectively define the energy landscape that a protein must navigate, influencing the stability of folding intermediates, the catalytic efficiency of chaperones, and the kinetics of disulfide bond formation [29] [61]. The subsequent sections will translate these principles into actionable, quantitative strategies and validated experimental workflows.
Scientific Background and Rationale
The Thermodynamic and Kinetic Framework of Refolding
Protein refolding is an inherently complex process governed by the principles of energy landscape theory, which envisions folding as a funnel-guided descent toward the native state, the global energy minimum [29] [61]. The cellular environment is replete with challenges such as molecular crowding, which increases the risk of aggregation by promoting non-specific intermolecular interactions [61]. Molecular chaperones, such as the E. coli GroEL/ES and Hsp70 (DnaK), function as essential components of the proteostasis network to mitigate these risks. They do not provide steric information for folding but instead act as catalysts that prevent off-pathway events, allowing the protein to explore its conformational landscape more efficiently [61] [62]. In vitro, the objective is to replicate this permissive environment by meticulously tuning the buffer conditions to favor productive intramolecular interactions over unproductive intermolecular ones.
The Critical Role of Chaperones in Shaping the Folding Landscape
Chaperones employ diverse mechanisms. ATP-independent "holdases" like Trigger Factor and small heat-shock proteins (sHsps) bind to nascent or stress-denatured polypeptides, preventing aggregation [25] [61] [62]. ATP-dependent "foldases" like Hsp70 and chaperonins undergo ATP-driven conformational cycles that can actively promote folding. For instance, the GroEL/ES system provides a sequestered, Anfinsen-like compartment that shields the folding protein from the crowded extracellular environment [61]. The efficacy of these chaperones is profoundly influenced by solution conditions. pH can alter their charge distribution and substrate affinity, temperature affects ATP hydrolysis rates and complex stability, and ionic strength can modulate electrostatic interactions critical for substrate binding and release [63] [61]. Understanding these dependencies is key to designing effective refolding buffers that leverage chaperone activity.
Optimization of Critical Parameters
The following section provides a detailed, parameter-wise breakdown for optimization, consolidating quantitative data for direct application.
Table 1: Optimization Parameters for Chaperone-Assisted Protein Refolding
| Parameter | Optimal Range | Mechanistic Role | Chaperone-Specific Considerations |
|---|---|---|---|
| pH | 7.0 - 8.5 | Influences charge state of amino acid side chains, stability of folding intermediates, and kinetics of disulfide bond formation. | Affects chaperone-substrate interactions; for example, the Hsp70 system is allosterically regulated by proline isomerization which can be pH-sensitive [61]. |
| Temperature | 4 - 25°C | Slows down aggressive hydrophobic collapse and aggregation kinetics, allowing for more controlled folding. | Lower temperatures (e.g., 10°C vs. 37°C) can increase the dependency of certain client proteins on the DnaK system [62]. |
| Ionic Strength | 50 - 200 mM NaCl | Modulates electrostatic interactions; low to moderate strength can shield repulsive charges without disrupting essential salt bridges. | High ionic strength can disrupt electrostatic steering mechanisms used by some chaperones for substrate recognition. |
| Redox Conditions | GSH:GSSG Ratio 5:1 to 10:1 | Provides a redox potential that facilitates thiol-disulfide exchange, enabling the formation of correct disulfide bonds. | Chaperones like Spy and GroEL function on non-disulfide bonded proteins; Dsb enzymes are required for disulfide bond formation in the periplasm. |
| Chaotrope (Urea) | 0.5 - 2.0 M | Suppresses aggregation by weakening hydrophobic interactions, keeping folding intermediates soluble. | Enables refolding of "chaperone-nonrefolders" like PGK in vitro, which otherwise require co-translational folding in vivo [62]. |
Protocol: Refolding by Direct Dilution
Principle: Rapidly reducing the concentration of a denaturant to a point where the protein can begin to fold, but where the presence of mild chaotropes or other additives suppresses aggregation [35].
Materials:
- Solubilized protein in high-concentration denaturant (e.g., 6 M GuHCl, 1-10 mM DTT).
- Refolding Buffer (See Table 1 for parameter ranges): Typically a 50-100 mM Tris or phosphate buffer, containing arginine, oxidized/reduced glutathione, and sub-denaturing concentrations of urea.
- Dialysis equipment or rapid mixing device.
Procedure:
- Solubilization and Reduction: Isolate and solubilize inclusion bodies in a denaturing buffer (e.g., 6 M GuHCl, 50 mM Tris, 1 mM EDTA, 100 mM DTT, pH 8.0) for 1-2 hours at room temperature [35].
- Clarification: Centrifuge the solubilized mixture at 15,000 × g for 20 minutes to remove any insoluble debris.
- Dilution: Rapidly dilute the clarified, denatured protein solution into a vigorously stirred refolding buffer. The typical protein concentration after dilution should be 10-100 µg/mL to minimize aggregation [35].
- Incubation: Allow the refolding reaction to proceed for 12-24 hours at a controlled temperature (e.g., 10°C).
- Concentration and Buffer Exchange: Concentrate the refolded protein using ultrafiltration and dialyze it into a final storage buffer to remove refolding additives.
Protocol: Chaperone-Assisted Refolding
Principle: Utilizing defined chaperone systems (e.g., GroEL/ES, DnaK/DnaJ/GrpE) to facilitate the folding of proteins that cannot refold spontaneously, mimicking the in vivo biogenesis pathway [61] [62].
Materials:
- Purified chaperone proteins (e.g., GroEL, GroES, DnaK, DnaJ, GrpE).
- ATP-regenerating system (e.g., ATP, phosphocreatine, creatine kinase).
- Refolding buffer without aggressive anti-aggregation agents that might interfere with chaperone-substrate interaction.
Procedure:
- Preparation of Unfolded Substrate: Denature the target protein in a high concentration of chaotope (e.g., 6 M GuHCl). Optionally, remove the reducing agent prior to refolding if disulfide bonds are required.
- Chaperone Addition: Dilute the denatured protein into a refolding buffer containing a pre-incubated mixture of the chaperone system. For GroEL/ES, a molar excess of GroEL to substrate (often 2:1 to 10:1) and a 2-10 fold excess of GroES to GroEL is typical [61] [62].
- Initiation of Folding: Add an ATP-regenerating system to initiate the chaperone cycle. For the DnaK system, include ATP and the co-chaperones DnaJ and GrpE.
- Incubation: Incubate the reaction for 1-2 hours at 25-37°C to allow for multiple rounds of the ATP-dependent chaperone cycle.
- Isolation of Folded Protein: Separate the folded target protein from the chaperones and aggregates using size-exclusion chromatography or native gel electrophoresis.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Refolding and Chaperone Studies
| Reagent | Function & Mechanism | Example Application |
|---|---|---|
| L-Arginine | A chemical chaperone that suppresses protein aggregation by weakening hydrophobic interactions without inhibiting refolding. | Used at 0.5 - 1.0 M in refolding buffers to increase productive yield [45] [35]. |
| GSH/GSSG Redox Pair | Establishes a redox potential that allows for thiol-disulfide exchange, catalyzing the formation of correct disulfide bonds. | A typical ratio is 5:1 to 10:1 (GSH:GSSG) at 1-5 mM total concentration [35]. |
| Glycerol & Sugars | Polyols that stabilize the native state of proteins via the preferential exclusion mechanism, shifting the equilibrium toward the folded form. | Used at 5-20% (v/v) glycerol or 0.5-1 M sucrose/sorbitol to enhance refolding yields [45]. |
| Trigger Factor (TF) | A ribosome-associated ATP-independent chaperone that binds nascent chains, preventing premature folding and aggregation during synthesis. | Studied in RNCs (Ribosome-Nascent Chain complexes) to understand co-translational folding pathways [25]. |
| GroEL/ES Chaperonin | Provides a central folding chamber, isolating a single polypeptide to fold in a protected, ATP-dependent manner. | Essential for refolding "chaperone-nonrefolder" proteins like MetK or RpoC in vitro [62]. |
Workflow and Pathway Visualization
The following diagram illustrates the critical decision points and pathways in a chaperone-assisted refolding experiment, integrating the parameters and reagents discussed.
Refolding Experiment Workflow
Chaperone-Assisted Folding Landscape
This diagram conceptualizes how chaperones reshape the protein folding energy landscape to prevent kinetic traps and guide the protein toward its native state.
Concluding Remarks
The optimization of pH, temperature, ionic strength, and redox conditions is a foundational activity in the development of robust chaperone-assisted refolding protocols. As the presented data and protocols underscore, there is no universal solution; success is achieved through the systematic and iterative screening of these parameters in the context of the target protein's specific biophysical characteristics. The integration of these classic biochemical principles with modern structural insights into chaperone mechanisms, such as those provided by techniques like READ [63] and HDX-MS [25], provides a powerful framework. This enables researchers to not only rescue recalcitrant proteins from inclusion bodies but also to fundamentally understand and control their journey back to a functional, native state.
Selecting the Right Chaperone System for Your Target Protein
Molecular chaperones are indispensable tools in modern biochemistry, preventing protein aggregation and facilitating the correct folding of substrate proteins (clients) into their native, functional states. The selection of an appropriate chaperone system is not trivial; it is a critical step that determines the success of protein refolding experiments, the production of recombinant proteins, and functional studies. The cellular proteostasis network is complex, comprising multiple chaperone families that operate in coordination with co-chaperones. This application note provides a structured framework, supported by quantitative data and detailed protocols, to guide researchers in selecting and implementing the optimal chaperone system for their specific target protein. Understanding that different chaperones have distinct client specificities—such as GroEL's preference for acidic, high molecular weight proteins and Hsp70's broad substrate range—forms the foundation for a rational selection process [64].
Major Chaperone Families and Their Mechanisms
The table below summarizes the key features of the major chaperone families commonly used in refolding assays.
Table 1: Key Features of Major Chaperone Families
| Chaperone Family | Representative Members | ATP-Dependent | Core Mechanism | Common Client Features |
|---|---|---|---|---|
| Hsp60/Chaperonins | GroEL/ES (E. coli), HSP60/HSP10 (human) | Yes | Forms an isolated folding cage; encapsulates clients for folding [64] | Acidic proteins (low pI), high molecular weight proteins, multi-domain proteins, α/β domain architectures [64] |
| Hsp70 | DnaK (E. coli), HSPA8 (human) | Yes | Binds short hydrophobic stretches in client peptides; cycle regulated by co-chaperones Hsp40 (DnaJ) and NEFs (GrpE) [65] | Broad range of clients; overlaps significantly with GroEL substrates [64] |
| Hsp90 | HtpG (E. coli), HSP90α/β (human) | Yes | Stabilizes conformation of metastable client proteins; involved in signal transduction [33] | Protein kinases, transcription factors, steroid hormone receptors [66] |
| Hsp100 | ClpB (E. coli), Hsp104 (yeast) | Yes | Disaggregates and reactivates aggregated proteins in concert with Hsp70 [67] | Aggregated proteins [67] |
| Small HSPs (sHSPs) | IbpA/B (E. coli), HSPB1 (HSP27, human) | No | First line of defense; binds unfolding clients to prevent aggregation [33] | Misfolded proteins with exposed hydrophobic surfaces [33] |
| ATP-Independent Holdases | Spy (E. coli) | No | Binds clients with weak affinity, allowing folding while chaperone-bound [60] [63] | Allows client folding while continuously bound; affinity must be optimized to avoid inhibiting folding [60] |
A Decision Framework for Chaperone Selection
The following diagram outlines a logical workflow for selecting a chaperone system based on the properties of your target protein and experimental goals.
Quantitative Data for Chaperone Selection
Proteome-wide studies in E. coli have quantified the refolding efficiency of different chaperone systems, providing a data-driven basis for selection. The following table summarizes the refolding capabilities of GroEL and the cytosolic environment, highlighting which structural classes of proteins are most dependent on chaperone assistance.
Table 2: Proteome-Wide Refolding Efficiency of GroEL and Cytosolic Milieu in E. coli [64]
| Protein Category | Refolding Efficiency in Cytosol (without dedicated chaperones) | Refolding Efficiency with GroEL/ES | Dependency on GroEL |
|---|---|---|---|
| Acidic Proteins (pI < 5) | Low | High (85% of detectable proteins refolded) | High |
| Basic Proteins (pI > 7) | High | Moderate improvement | Low |
| High Molecular Weight Proteins | Low | High | High |
| Proteins with Many Domains | Low | High | High |
| α/β Domain Architectures | Low | High | High |
| Cohort of Intransigent Proteins | Cannot refold | Cannot refold | N/A (May require cotranslational folding) |
Key insights from this data include:
- GroEL is crucial for acidic proteins: 85% of the acidic proteins for which data was available were refolded by GroEL, whereas they refolded poorly in the cytosolic milieu alone [64].
- Basic proteins are efficient spontaneous refolders: These proteins often refold efficiently without dedicated chaperone systems, though the cytosolic environment itself provides beneficial factors [64].
- Overlap between GroEL and DnaK substrates: Despite vastly different structures and mechanisms, GroEL (Hsp60) and DnaK (Hsp70) can refold a similar set of proteins, suggesting a common mechanism of unfolding misfolded states [64].
Detailed Experimental Protocols
Protocol: Spontaneous and GroEL/ES-Assisted Refolding of sGFP
This protocol uses a slow-folding mutant of GFP (sGFP), which is non-fluorescent when unfolded and fluoresces upon reaching its native state, allowing real-time monitoring of refolding kinetics [47].
I. Research Reagent Solutions
Table 3: Essential Reagents for sGFP Refolding Assay [47]
| Reagent | Function/Description |
|---|---|
| sGFP (Slow-folding GFP mutant) | Model substrate protein; fluorescence indicates correct folding. |
| Guanidine Hydrochloride (GuHCl) | Chemical denaturant used to unfold the substrate protein. |
| GroEL (Hsp60) | Core chaperonin; forms a tetradecameric barrel. |
| GroES (Hsp10) | Co-chaperonin; acts as a "lid" for the GroEL barrel. |
| Adenosine Triphosphate (ATP) | Energy source to drive the chaperonin folding cycle. |
| Buffer A (Refolding Buffer) | Provides a folding-favorable environment (Typically: 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 10 mM MgCl₂). |
| Dithiothreitol (DTT) | Reducing agent to prevent disulfide bond formation. |
II. Step-by-Step Procedure
Protein Unfolding:
- Combine 2.5 µL of 20 µM native sGFP with 7.5 µL of 8 M GuHCl in Buffer A. The final GuHCl concentration in the unfolding reaction is 6 M.
- Incubate the mixture for 1 hour at 25°C to achieve complete unfolding [47].
Instrument Setup:
- Switch on a fluorescence spectrofluorometer and allow the lamp to warm up for 45 minutes to minimize fluctuations.
- Set the instrument to kinetic acquisition mode with the following parameters:
- Excitation wavelength: 480 nm
- Emission wavelength: 515 nm
- Temperature: 25°C
- Total kinetics time: 1,800 seconds (30 minutes) [47].
Spontaneous Refolding (Control):
- Dilute 2 µL of the unfolded sGFP mixture 100-fold into 198 µL of Buffer A in a fluorescence cuvette (final sGFP concentration ~200 nM). Mix thoroughly by pipetting.
- Immediately begin kinetic data acquisition to monitor the increase in fluorescence at 515 nm over time [47].
Chaperonin-Assisted Refolding:
- Dilute 2 µL of the unfolded sGFP mixture 100-fold into 198 µL of Buffer A that has been pre-supplemented with:
- 400 nM tetradecamers of GroEL
- 800 nM heptamers of GroES
- 2 mM ATP (final concentration)
- The recommended molar ratio is 1 sGFP : 2 GroEL : 4 GroES.
- Mix thoroughly and start kinetics acquisition immediately [47].
- Dilute 2 µL of the unfolded sGFP mixture 100-fold into 198 µL of Buffer A that has been pre-supplemented with:
Data Analysis:
- The refolding kinetics trace is typically best fitted by a single exponential equation:
y = y₀ + A₁ * e^(-x/t₁)wherey₀is the offset,A₁is the amplitude, andt₁is the time constant. - The apparent refolding rate is the reciprocal of the time constant (1/t₁) [47].
- The refolding kinetics trace is typically best fitted by a single exponential equation:
Protocol: Assessing Refolding via Limited Proteolysis-Mass Spectrometry (LiP-MS)
For proteome-wide or complex refolding assessments, LiP-MS is a powerful tool that monitors structural changes by measuring protease accessibility.
Key Steps for LiP-MS [64]:
- Sample Preparation: Create a native lysate (control) and a denatured lysate (unfolded in 6 M guanidinium chloride).
- Refolding: Rapidly dilute the denatured lysate into a refolding buffer (e.g., cytosolic medium or a defined buffer) with or without the chaperone system of interest.
- Limited Proteolysis: After a refolding period, treat all samples with a nonspecific protease like Proteinase K (PK). PK cleaves proteins in solvent-exposed and flexible regions, which are more abundant in misfolded states.
- Analysis: Use LC-MS/MS to sequence and quantify the resulting peptide fragments. Compare the peptide profiles of the refolded samples to the native control. Proteins that have not refolded correctly will show different proteolytic patterns than the native control, revealing their refolding status and chaperone dependence [64].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Research Reagents for Chaperone Studies
| Reagent / Material | Function in Chaperone-Assisted Refolding |
|---|---|
| GroEL/ES (Hsp60/Hsp10) | The classic chaperonin system; essential for refolding acidic, high MW, and multi-domain proteins in an ATP-dependent manner [64] [47]. |
| DnaK/DnaJ/GrpE (Hsp70/Hsp40/NEF) | The major Hsp70 system; DnaK is the core chaperone, DnaJ targets clients and stimulates ATPase activity, GrpE promotes nucleotide exchange for client release [64] [65]. |
| sGFP (Slow-folding GFP) | An ideal fluorescent model substrate for real-time monitoring of refolding kinetics in vitro [47]. |
| Spy (E. coli) | An ATP-independent chaperone; valuable for studying "folding-while-bound" mechanisms and the role of binding affinity on folding efficiency [60] [63]. |
| Cyto-Serum | A reconstituted cytosolic medium containing physiological concentrations of metabolites, ions, and cofactors; used to study refolding in a near-native environment [64]. |
| 4-Iodophenylalanine (pI-Phe) | A non-canonical amino acid used for anomalous scattering in X-ray crystallography (e.g., READ method) to visualize dynamic chaperone-substrate complexes [63]. |
| Proteinase K | A nonspecific protease used in Limited Proteolysis (LiP) assays to probe the structural states of proteins during refolding [64]. |
The Role of Co-chaperones and Ligands in Enhancing Folding Efficiency
Within the intricate cellular environment, the journey of a polypeptide chain to its functional three-dimensional structure is a complex process facilitated by the proteostasis network—an integrated system of molecular chaperones, folding enzymes, and degradation machineries [3]. Central to this network are molecular chaperones such as Hsp70, Hsp90, and chaperonins, which prevent protein aggregation and assist in proper folding [68]. However, the efficient functioning of these chaperones is often dependent on co-chaperones and ligands that regulate their activity and specificity.
Co-chaperones are specialized proteins that physically associate with core chaperones to modulate their function, client binding, and ATPase cycles [41]. These regulatory components, along with chemical ligands, are indispensable for managing the folding of a diverse clientele, from nascent chains to complex regulatory proteins. This application note examines the mechanisms by which co-chaperones and ligands enhance folding efficiency, providing detailed protocols and analytical frameworks for researchers investigating chaperone-assisted protein refolding with applications in basic research and drug development.
Co-chaperone Mechanisms and Client Specificity
Co-chaperones perform specialized functions within the chaperone network, often acting as adaptors that determine substrate specificity or modulate the ATP-dependent conformational changes of their partner chaperones.
FKBP4-Hsp90 Complex in CCT8 Folding
FKBP4 is a co-chaperone of Hsp90 with increased expression in multiple types of cancers [68]. Recent research employing proximity-dependent biotin identification (BioID) mass spectrometry revealed that the FKBP4-Hsp90 complex plays a critical role in the folding of CCT8, a subunit of the chaperonin containing TCP-1 (CCT) complex. Analysis of BioID mass spectrometry data demonstrated that cadherin-binding proteins represent the top category of FKBP4-associated proteins, with CCT8 identified among these interactions [68].
Table 1: Functional Roles of Select Co-chaperone Systems
| Co-chaperone | Partner Chaperone | Key Function | Client Protein Example |
|---|---|---|---|
| FKBP4 | Hsp90 | Facilitates folding of specific substrates | CCT8 (CCT complex subunit) |
| Hop (STIP1) | Hsp70/Hsp90 | Bridges chaperone complexes; facilitates client transfer | Glucocorticoid receptor (GR) |
| PPIases (e.g., Cyclophilins, FKBPs) | Hsp90, Hsp70 | Catalyze proline isomerization; allosteric regulation | Ribonuclease A (RNase A); Hsp70 ATPase domain |
| P23 | Hsp90 | Stabilizes mature client complexes | Ligand-bound glucocorticoid receptor |
Functional validation through knockdown experiments confirmed the biological significance of this interaction, as reduction of FKBP4 led to the aggregation of CCT8 and compromised the stability of CCT8 clients, including CDK2 and α-tubulin [68]. This finding demonstrates a functional crosstalk between two essential protein folding systems—the FKBP4-Hsp90 complex and the CCT chaperonin—highlighting the interconnected nature of the cellular folding machinery.
Hsp70-Hsp90 Coordinating Co-chaperones
The handover of clients between Hsp70 and Hsp90 systems is mediated by the co-chaperone Hop (Hsp70/Hsp90-organizing protein), which facilitates the assembly of intermediate chaperone complexes [41]. This coordinated action is particularly evident in the maturation of the glucocorticoid receptor (GR), where Hop participates in the formation of an "loading complex" with Hsp70 and Hsp90, while p23 associates with the final "maturation complex" containing Hsp90 [41]. This sequential involvement of different co-chaperones ensures proper folding and activation of signaling proteins.
Peptidyl Prolyl Isomerases (PPIases) as Catalytic Co-chaperones
PPIases, including cyclophilins and FK506-binding proteins (FKBPs), catalyze the slow interconversion between cis and trans configurations of proline, a rate-limiting step in the folding of many proteins [41]. Beyond their enzymatic activity, these proteins also function as co-chaperones by interacting with Hsp90 through tetratricopeptide repeat (TPR) domains, with this interaction being essential for steroid hormone receptor complexes [41]. Some PPIases also allosterically regulate chaperone function; for instance, Hsp70 is proposed to be regulated by isomerization at Pro143, a residue critical for stabilizing the open conformation of its substrate-binding pocket [41].
Quantitative Analysis of Chaperone Efficacy
Systematic studies have quantified the impact of chaperones and co-chaperones on protein folding efficiency, revealing key structural determinants of chaperone dependency.
Proteome-Wide Refolding Efficiency
A recent proteome-wide study using limited proteolysis-mass spectrometry (LiP-MS) to monitor refolding of E. coli proteins revealed that GroEL/ES can refold approximately 85% of proteins for which data were obtainable [64]. The study identified distinct protein characteristics that correlate with chaperone dependency:
Table 2: Protein Features Correlating with Chaperone Dependency
| Protein Feature | Impact on Refolding | Primary Assisting Chaperone |
|---|---|---|
| Acidic pI | Increased dependency | GroEL/ES |
| High Molecular Weight | Increased dependency | GroEL/ES |
| Multi-domain Architecture | Increased dependency | GroEL/ES |
| α/β Domain Architecture | Increased dependency | GroEL/ES |
| Basic pI | Efficient independent refolding | Minimal chaperone requirement |
The research demonstrated that acidic proteins show greater reliance on GroEL for successful refolding, while basic proteins generally refold efficiently without chaperone assistance [64]. This systematic approach provides a map for predicting which types of proteins are more reliant on chaperones and has revealed that DnaK and GroEL refold a largely overlapping set of proteins, suggesting a common mechanism despite their vastly different structures [64].
Experimental Validation of Co-chaperone Impact
The essential nature of co-chaperones is evident from functional studies showing that:
- FKBP4 knockdown directly causes CCT8 aggregation [68]
- Compromised stability of CCT8 clients (CDK2 and α-tubulin) upon FKBP4 reduction [68]
- Catalytic proline isomerization by PPIases can be rate-limiting for folding [41]
Application Notes: Experimental Protocols
Differential Scanning Fluorimetry Guided Refolding (DGR) Protocol
The DGR approach provides a high-throughput method for identifying optimal refolding conditions for proteins recovered from inclusion bodies [69].
Protein Denaturation and Primary Screen
- Isolation of Inclusion Bodies: Resuspend cell pellets in lysis buffer (50 mM Tris-HCl, pH 8.0; 300 mM NaCl; 5% glycerol; 3 mM β-mercaptoethanol; 5 mM EDTA). Perform repeated sonication and centrifugation cycles with 0.5% Triton X-100 to purify inclusion bodies [69].
- Solubilization/Denaturation: Resuspend inclusion bodies in either 8 M urea or 6 M guanidine-HCl. Remove insoluble material by centrifugation at 19,000 × g for 20 minutes [69].
- Primary pH Refolding Screen:
- Prepare a 96-well plate with 190 μL of refolding buffers covering various pH conditions.
- Add 10 μL of denatured protein solution (~5 mg/mL) to each well (1:20 dilution).
- Incubate with gentle shaking at room temperature or 4°C.
- Analyze refolding at multiple time points to identify optimal incubation duration [69].
Detection and Analysis
- Differential Scanning Fluorimetry:
- Combine 5 μL of 25× diluted SYPRO Orange with 45 μL of refolding sample.
- Perform thermal denaturation from 22°C to 90°C with increments of 0.5°C per 30 seconds.
- Monitor fluorescence (Excitation: 485 nm, Emission: 530 nm) [69].
- Data Interpretation: Shift in melting temperature (Tm) indicates successful refolding, distinguishing properly folded proteins from misfolded aggregates.
Secondary Screen and Scale-up
- Secondary Additive Screen: Test "helper" molecules including arginine, chaotropes (urea), stabilizing agents, counter-ions, and redox couples.
- Scale-up Refolding: Add 2.5 mL denatured protein dropwise to 50 mL optimized refolding buffer with continuous stirring.
- Dialysis Buffer Optimization: Use thermal shift assay to identify optimal buffers for removing refolding agents [69].
Chaperone Co-expression Protocol for Improved Solubility
For enhancing soluble yield of recombinant proteins in E. coli:
- Co-expression System: Utilize eight combinations of molecular chaperones (e.g., DnaK/DnaJ/GrpE/ClpB) co-expressed with the target recombinant protein [70].
- Two-step Protocol:
- Phase 1: Co-accumulate molecular chaperones and target protein.
- Phase 2: Disaggregate and refold target protein aggregates after blocking protein synthesis [70].
- Small-scale Purification: Perform screening of expression combinations before scaling up production. This approach has increased solubility for 70% of tested constructs with yields up to 42-fold higher than controls [70].
Visualization of Chaperone-Co-chaperone Workflows
Figure 1: Chaperone and Co-chaperone Coordination in Protein Folding
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Chaperone Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Molecular Chaperones | GroEL/ES, DnaK/DnaJ/GrpE, Hsp90, TRiC/CCT | Core folding machinery for in vitro refolding studies |
| Co-chaperones | FKBP4, Hop (STIP1), p23, PPIases (Cyclophilins, FKBPs) | Regulate chaperone cycle and client specificity |
| Chemical Chaperones | Arginine, Glycerol, Betaine | Reduce aggregation during refolding |
| Denaturants | Urea (8 M), Guanidine-HCl (6 M) | Solubilize inclusion bodies |
| Detection Reagents | SYPRO Orange, ANS | Monitor folding state via fluorescence |
| Redox Couples | GS SG / GSH, Cysteine/Cystamine | Regulate disulfide bond formation |
| Commercial Kits | QuickFold, FoldIt, iFOLD | Pre-formulated refolding condition screens |
Co-chaperones and ligands serve as essential regulatory components that enhance folding efficiency by modulating chaperone specificity and catalytic function. The experimental protocols and analytical frameworks presented here provide researchers with robust methods for investigating and manipulating these systems. The growing understanding of how co-chaperones like FKBP4 interface with major chaperone networks opens new avenues for therapeutic interventions targeting proteostasis in diseases such as cancer and neurodegeneration. As research continues to elucidate the intricate coordination within the chaperone network, the ability to rationally manipulate folding pathways for research and therapeutic purposes will continue to advance.
This application note provides a detailed mechanistic comparison of artificial-chaperone-assisted and unassisted refolding of urea-denatured carbonic anhydrase B (CAB). The data demonstrate that while artificial chaperones significantly improve the final yield of active enzyme by preventing aggregation, they do not alter the fundamental rate-determining step of the folding process itself. The assisted protocol, utilizing a detergent and cyclodextrin pair, is highly effective under conditions where traditional dilution methods fail, making it particularly valuable for recovering bioactive proteins from inclusion bodies.
The production of recombinant proteins in systems like E. coli is frequently hampered by the formation of inclusion bodies—insoluble aggregates of misfolded protein. In vitro refolding is therefore a critical step in obtaining functional protein for research and therapeutic applications. However, refolding is often inefficient due to competition between productive folding and off-pathway aggregation [71]. This case study focuses on the refolding of Bovine Carbonic Anhydrase B (CAB), a model enzyme notoriously prone to aggregation during refolding [72].
We have previously described an artificial chaperone system—a biomimetic strategy inspired by natural chaperone proteins like GroEL/GroES [71]. This system uses a pair of low molecular weight folding assistants: a detergent to capture the unfolded protein and prevent aggregation, and cyclodextrin to strip the detergent away, inducing folding [42] [73]. This note presents a direct mechanistic comparison of this assisted method against unassisted dilution refolding, providing both quantitative data and reproducible protocols.
Key Experimental Findings & Quantitative Data
Comparative Refolding Yields and Rates
The core finding is that artificial chaperones dramatically enhance the recovery of active CAB without changing the intrinsic rate-limiting step of its folding pathway [74].
Table 1: Summary of Refolding Outcomes for CAB under Different Conditions
| Refolding Condition | Final Yield of Active Enzyme | Impact on Aggregation | Rate-Determining Step |
|---|---|---|---|
| Unassisted Refolding | Low to negligible [42] [73] | High aggregation | Slow partial folding step, indistinguishable from assisted refolding [74] |
| Artificial Chaperone-Assisted | Excellent (high yield) [42] [73] | Effectively prevented [42] | Same as unassisted refolding [74] |
| Slow Detergent Removal (Dialysis) | Less effective [73] | Not applicable | Dependent on stripping kinetics [73] |
The data indicate a distinctive product-determining step early in the assisted mechanism, where the formation of a productive protein-detergent complex dictates the final yield. However, once a competent complex is formed and the detergent is stripped, the subsequent folding to the native state proceeds through the same slow, rate-limiting step as in unassisted refolding [74].
Essential Reagent Solutions for Artificial Chaperone Refolding
The successful implementation of this protocol relies on specific reagents, each playing a critical role in the two-step mechanism.
Table 2: Key Research Reagent Solutions for Artificial Chaperone Refolding
| Reagent | Function in Protocol | Key Mechanistic Role | Examples/Notes |
|---|---|---|---|
| Detergents | Captures unfolded protein during dilution. | Forms a complex with hydrophobic regions of the non-native protein, shielding them and preventing intermolecular aggregation [42] [73]. | Can be anionic or cationic (e.g., CTAB) [74]. Effective even at sub-micellar concentrations [73]. |
| Cyclodextrins | Strips detergent from the protein-detergent complex. | Its hydrophobic cavity encapsulates and removes detergent molecules, allowing the protein to refold in a controlled manner [42] [73]. | Rapid stripping is crucial for high efficiency [73]. |
| Denaturants | Unfolds the target protein from inclusion bodies. | Disrupts non-covalent interactions to solubilize protein aggregates. | Guanidinium HCl or Urea [42] [74]. |
| Folding Buffers | Provides the correct chemical environment for renaturation. | Controls pH, ionic strength, and redox potential to favor native structure formation. | May contain additives like glycerol or PEG [72]. |
Experimental Protocols
Protocol A: Artificial Chaperone-Assisted Refolding of CAB
This protocol is adapted from the detailed studies of Rozema & Gellman and Hanson & Gellman [42] [73].
Principle: Unfolded CAB is first captured by a detergent to form a non-aggregated, folding-competent intermediate. The subsequent addition of cyclodextrin removes the detergent, inducing the protein to refold into its native, active conformation.
Materials:
- Denatured CAB (e.g., in 6 M GuHCl or 8 M Urea).
- Capture Detergent (e.g., Cetyltrimethylammonium bromide, CTAB).
- Stripping Agent (e.g., β-Cyclodextrin).
- Folding Buffer (e.g., 50 mM Tris-HCl, pH 7.5-8.0).
- Equipment: Standard lab equipment (pipettes, vortex, water bath).
Procedure:
- Denatured Protein Preparation: Prepare CAB denatured in 6 M GuHCl or 8 M urea at a concentration of approximately 1-2 mg/mL.
- Capture Step: Dilute the denatured CAB solution 50-100 fold into Folding Buffer pre-supplemented with the capture detergent (e.g., 1-5 mM CTAB). Mix gently but thoroughly.
- Critical Note: The detergent captures the unfolded protein immediately upon dilution, forming a complex that prevents aggregation.
- Incubation: Hold the mixture at the desired refolding temperature (e.g., 25°C) for 5-10 minutes.
- Stripping & Refolding Step: Introduce the stripping agent, β-cyclodextrin, to the above mixture. Use a molar excess of cyclodextrin relative to the detergent (e.g., a 2:1 molar ratio of cyclodextrin to detergent).
- Critical Note: Rapid and efficient stripping of the detergent is essential for high refolding yields. Slow methods like dialysis are less effective [73].
- Final Incubation: Allow the refolding reaction to proceed for a sufficient time (e.g., 1-2 hours or overnight) to reach completion.
- Analysis: Monitor protein refolding and quantify the yield of active enzyme using an activity assay (e.g., esterase activity for CAB) and analyze aggregation by size-exclusion chromatography or native PAGE.
The workflow for this protocol is illustrated below.
Protocol B: Unassisted (Dilution) Refolding of CAB
This protocol serves as a control to highlight the efficiency of the artificial chaperone system.
Principle: The denaturant is diluted to a non-denaturing concentration, relying on the intrinsic properties of the protein to spontaneously refold, a process often hampered by aggregation.
Materials:
- Denatured CAB (identical to Protocol A).
- Folding Buffer (identical to Protocol A, but without additives).
Procedure:
- Denatured Protein Preparation: Use the same stock of denatured CAB as in Protocol A.
- Dilution Refolding: Dilute the denatured CAB solution 50-100 fold into Folding Buffer lacking any detergent or cyclodextrin. Mix gently.
- Incubation: Allow the solution to incubate for the same duration as in Protocol A.
- Analysis: Quantify active enzyme and aggregation using the same methods as Protocol A. Expect significantly lower yields of active protein and visible precipitation or high molecular weight aggregates [42] [73].
Mechanistic Insights and Discussion
The comparative data lead to a clear mechanistic model for the artificial chaperone system. The following diagram summarizes the pathways and key mechanistic insights.
Key Interpretations:
- Aggregation Prevention is Key: The primary mechanism by which artificial chaperones enhance yield is by physically blocking aggregation. The detergent acts as a shield for exposed hydrophobic patches on the folding intermediates, which would otherwise cause irreversible association [42] [73].
- Fidelity of the Native Pathway: The observation that the rate-determining step is identical in both assisted and unassisted refolding is critical [74]. It indicates that the artificial chaperone system does not force the protein down a novel folding pathway. Instead, it prevents off-pathway aggregation, allowing a greater proportion of molecules to proceed correctly through the intrinsic, albeit slow, folding pathway.
- Kinetics of Stripping: The requirement for rapid detergent removal by cyclodextrin, as opposed to slow dialysis, highlights that the protein-detergent complex, while preventing aggregation, is a kinetic trap. Efficient escape from this trap is necessary to feed molecules into the productive folding funnel [73].
The artificial chaperone-assisted refolding protocol provides a robust and highly effective method for recovering active carbonic anhydrase B from denatured states where conventional dilution methods fail. Its two-step capture-and-release mechanism effectively suppresses aggregation, the major competitor to productive folding. This mechanistic understanding and the associated protocols are directly applicable to the challenge of refolding other aggregation-prone recombinant proteins of scientific and therapeutic interest.
Assessing Fidelity and Function: Validation and Comparative Analysis
Within the framework of chaperone-assisted protein refolding research, functional assays are indispensable for quantifying the success of refolding protocols by measuring the recovery of a protein's native biological activity. The return of enzymatic function is a definitive indicator that a protein has adopted its correct three-dimensional structure. Fluorescence-based kinetic assays, in particular, have become a cornerstone technique due to their high sensitivity, real-time monitoring capabilities, and compatibility with high-throughput screening (HTS) formats [75] [76]. These assays are crucial for characterizing the folding journey of client proteins, from their initial interaction with chaperones like Hsp70 and Hsp40 to their final, functionally active state [41] [77]. This document provides detailed application notes and structured protocols for employing these powerful assays in the context of protein refolding research.
The Role of Functional Assays in Protein Refolding Research
Protein folding is a fundamental process essential for cellular health, yet it is prone to errors that can lead to misfolding and aggregation. The cellular proteostasis network, comprising molecular chaperones and folding enzymes, has evolved to guide newly synthesized polypeptides toward their native structures [41] [3]. Molecular chaperones such as Hsp70 and Hsp90 are ATP-dependent machines that prevent off-pathway misfolding and aggregation by binding to unfolding intermediates [41] [4].
A key challenge in this field is the fundamental disconnect observed between a protein's refolding behavior in a simplified test tube environment and its folding pathway during biosynthesis in the complex cellular milieu. Recent research using limited proteolysis-mass spectrometry (LiP-MS) has revealed that many proteins which cannot refold after chemical denaturation in vitro achieve native-like structure during co-translational synthesis in the cell without the aid of chaperones [78]. This underscores the critical importance of studying folding and refolding in a context that is as biologically relevant as possible.
Functional assays bridge this gap by providing a quantitative readout of successful folding. The regaining of enzymatic activity signifies that the protein has not only adopted a compact structure but has also formed the precise active site geometry required for catalysis. This is especially important for evaluating the efficacy of chaperone systems in refolding their client proteins, as the ultimate goal is to produce a functional enzyme, not just a soluble one.
Fluorescence-Based Kinetic Assays: Principles and Applications
Fluorometric assays have become essential tools in enzymatic studies and drug discovery due to their high sensitivity, specificity, and capacity for real-time monitoring [75] [76]. The underlying principle involves the use of a substrate that, upon enzymatic conversion, produces a fluorescent product. The increase in fluorescence intensity over time is directly proportional to the enzyme's activity, allowing researchers to determine kinetic parameters such as the Michaelis constant ((Km)) and the maximum reaction rate ((V{max})).
These assays are particularly well-suited for several applications in refolding research:
- High-Throughput Screening (HTS): Fluorescence-based formats are easily automated and miniaturized, making them ideal for rapidly screening large libraries of compounds for chaperone modulators or for identifying conditions that promote the refolding of intractable client proteins [75].
- Mechanistic Studies: By providing real-time kinetic data, these assays can reveal the mechanism of action of chaperones. For instance, they can help determine whether a chaperone simply prevents aggregation (a holdase) or actively accelerates the folding rate (a foldase) [41].
- Studying Endocannabinoid Hydrolytic Enzymes: As a specific example, fluorescence-based assays are crucial for understanding the activity and inhibition of enzymes like fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), which are important for maintaining endocannabinoid homeostasis [75].
The following diagram illustrates the logical workflow for developing and applying a fluorescence-based activity assay in a refolding context.
Essential Reagents and Instrumentation
A successful fluorescence-based refolding assay relies on a core set of reagents and instruments. The table below catalogues key components of the "scientist's toolkit" for this field.
Table 1: Research Reagent Solutions for Fluorescence-Based Refolding Assays
| Item | Function/Description | Application in Refolding |
|---|---|---|
| Fluorogenic Substrate | A non-fluorescent molecule that is converted into a highly fluorescent product by the target enzyme. | Serves as the direct reporter of enzymatic activity recovery during refolding [75]. |
| Molecular Chaperones | Proteins like Hsp70, Hsp40 (DnaK, DnaJ), and GroEL/ES that assist protein folding. | The central object of study; their ability to refold denatured client proteins is quantified [41] [78]. |
| ATP Regeneration System | A mixture (e.g., ATP, creatine phosphate, creatine kinase) that maintains constant ATP levels. | Essential for the function of ATP-dependent foldase chaperones like Hsp70 and GroEL [41]. |
| Refolding Buffer | A carefully optimized buffer that may include salts, redox agents, and crowding agents. | Provides the physico-chemical environment conducive to protein refolding and prevents aggregation [4]. |
| Microplate Reader | An instrument capable of measuring fluorescence intensity in multi-well plates with temperature control. | Enables high-throughput kinetic measurements of multiple refolding reactions simultaneously [76]. |
| Stopped-Flow Instrument | A device for rapidly mixing solutions and initiating reactions on millisecond timescales. | Used for measuring very fast folding and enzymatic kinetics that occur immediately after refolding initiation [79]. |
Quantitative Data from Functional Refolding Assays
The quantitative output from fluorescence-based refolding assays provides deep insights into chaperone function and folding efficiency. Data can be tabulated for clear comparison across different experimental conditions, such as the presence or absence of specific chaperones or co-factors.
Table 2: Exemplary Kinetic Data from a Chaperone-Assisted Refolding Experiment
| Refolding Condition | Final Activity Recovery (%) | Apparent (K_m) (µM) | Apparent (k_{cat}) (s⁻¹) | Lag Phase (min) |
|---|---|---|---|---|
| Spontaneous Refolding | 25 ± 5 | 150 ± 20 | 0.5 ± 0.1 | 15.0 ± 2.0 |
| + Hsp70 (DnaK) only | 40 ± 6 | 130 ± 15 | 0.8 ± 0.1 | 10.5 ± 1.5 |
| + Hsp70 & Hsp40 (DnaKJ) | 75 ± 8 | 95 ± 10 | 1.9 ± 0.2 | 2.0 ± 0.5 |
| + GroEL/ES System | 85 ± 5 | 90 ± 8 | 2.1 ± 0.2 | 1.5 ± 0.5 |
Note: The data is illustrative, based on common trends observed in refolding literature [41] [78]. The presence of the full chaperone system (DnaK/J or GroEL/ES) significantly enhances the yield and rate of functional enzyme recovery.
Detailed Protocol: Monitoring Chaperone-Assisted Refolding via Fluorescence Kinetics
This protocol outlines the steps to assess the refolding of a denatured model enzyme, such as a phosphatase or kinase, facilitated by the Hsp70/Hsp40 chaperone system, using a fluorogenic substrate.
Materials and Reagents
- Purified Proteins: Client enzyme (e.g., CheY [79]), Hsp70 (DnaK), Hsp40 (DnaJ), and a nucleotide exchange factor (GrpE).
- Denaturation Buffer: 6 M Guanidine-HCl, 50 mM Tris-HCl pH 8.0, 5 mM DTT.
- Refolding Buffer: 50 mM HEPES-KOH pH 7.5, 50 mM KCl, 10 mM MgCl₂, 2 mM DTT.
- ATP Solution: 100 mM ATP, pH 7.0, prepared in refolding buffer.
- Fluorogenic Substrate: A substrate specific to the client enzyme (e.g., a phosphorylated peptide conjugated to a fluorophore like MCA or AMC).
- Instrument: Fluorescence microplate reader with temperature control (capable of kinetic measurements).
Experimental Procedure
- Denaturation: Dilute the purified client enzyme into denaturation buffer to a final concentration of 10 µM. Incubate for 60 minutes at 25°C to ensure complete unfolding.
- Initiate Refolding:
- Prepare the refolding master mix in refolding buffer on ice. For a 100 µL final reaction volume:
- 5 µM Hsp70 (DnaK)
- 1 µM Hsp40 (DnaJ)
- 1 µM GrpE
- 2 mM ATP
- Rapidly dilute the denatured client enzyme 100-fold into the pre-chilled master mix to initiate refolding (final client enzyme concentration: 100 nM). Mix quickly but gently.
- Prepare the refolding master mix in refolding buffer on ice. For a 100 µL final reaction volume:
- Kinetic Measurement:
- Immediately transfer 100 µL of the refolding reaction to a black 96-well plate.
- Place the plate in the pre-warmed (25°C) microplate reader.
- Initiate kinetic fluorescence measurements (e.g., Ex/Em = 360/460 nm), reading the plate every 30 seconds for 60-120 minutes.
- Control Reactions:
- Negative Control: Perform refolding in the absence of chaperones and ATP.
- Background Control: Include a reaction with all components except the fluorogenic substrate to account for any background fluorescence.
- Termination & Calibration:
- After the kinetic read, add a known amount of native, fully active client enzyme to one well to obtain a fluorescence value corresponding to 100% activity.
- Alternatively, generate a standard curve with the native enzyme.
Data Analysis
- Subtract the background fluorescence from all kinetic traces.
- Normalize the fluorescence data to the maximum signal from the 100% activity control.
- Plot normalized activity (%) versus time to generate refolding progress curves for each condition.
- Compare the final yield of active enzyme and the kinetics of activity recovery between spontaneous and chaperone-assisted refolding.
The following workflow diagram maps the key experimental steps and the chaperone-client interactions that underpin the assay.
Advanced Applications: Co-Translational Folding Assays
While refolding from full denaturation is a classic approach, it is now evident that a protein's folding in vivo is predominantly co-translational, beginning as the polypeptide chain emerges from the ribosome [28]. Advanced methods like Arrest Peptide Profiling (AP Profiling) have been developed to quantitatively define these co-translational folding pathways in live cells with codon resolution [28].
This high-throughput method uses a force-sensing arrest peptide (SecM) from E. coli inserted into a gene of interest. When the nascent protein domain folds co-translationally, it generates mechanical force that pulls on the arrest peptide, leading to the release of translational arrest and the expression of a downstream reporter (e.g., GFP). The level of reporter expression is directly correlated with the folding efficiency of the nascent chain at that specific length. This allows researchers to map precisely when and where a protein domain folds during its synthesis and how different chaperones facilitate this process [28].
Such methods are revolutionizing the field by providing an unprecedented, high-resolution view of folding in its native cellular context, bridging the gap between traditional in vitro refolding assays and complex cellular physiology.
The study of protein folding, particularly chaperone-assisted refolding, relies heavily on advanced biophysical techniques to monitor structural changes, conformational dynamics, and folding pathways. Hydrogen-deuterium exchange mass spectrometry (HDX-MS), various spectroscopic methods, and chromatographic techniques have emerged as powerful tools for providing high-resolution insights into protein folding mechanisms. These methods enable researchers to move beyond simple activity assays and examine the intricate structural transitions that occur as proteins navigate their energy landscapes toward native conformations, often with the assistance of molecular chaperones like GroEL/ES, Hsp70, and Hsp90 [80] [41].
Within the complex cellular environment, molecular chaperones play indispensable roles in assisting protein folding, preventing aggregation, and remodeling misfolded states. Understanding their mechanisms requires techniques capable of capturing transient intermediates and mapping binding interfaces. HDX-MS has proven particularly valuable in this context, revealing how chaperones interact with client proteins, induce allosteric conformational changes, and participate in coordinated folding cycles [80]. When combined with spectroscopic and chromatographic approaches, these methods form a comprehensive toolkit for validating protein refolding protocols and elucidating the fundamental principles of chaperone-assisted folding.
HDX-MS for Chaperone-Assisted Refolding Analysis
Principle and Application
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) measures the exchange rate between backbone amide hydrogens in proteins and deuterium atoms from the solvent. This exchange rate is exquisitely sensitive to protein folding状态; amide hydrogens involved in stable hydrogen bonding or buried within the protein core exchange slowly, while those in unstructured or dynamic regions exchange rapidly. By monitoring deuterium incorporation over time, HDX-MS provides a detailed map of protein conformational dynamics and stability at peptide-level resolution [80] [81].
The application of HDX-MS to chaperone-assisted protein folding has revealed several key mechanisms. Studies on GroEL/GroES, Hsp70, and Hsp90 have shown that these chaperones undergo specific conformational changes during their functional cycles, which can be precisely mapped using HDX-MS. The technique has also been instrumental in identifying chaperone-client binding interfaces and understanding how allosteric signals are communicated through chaperone networks. Furthermore, HDX-MS has illuminated how chaperones remodel client proteins by stabilizing folding intermediates and preventing off-pathway aggregation [80].
Technical Workflow and Protocol
The standard HDX-MS protocol for analyzing chaperone-assisted refolding involves several critical steps that must be carefully controlled to ensure reproducible results. The following workflow diagram illustrates the key stages in the HDX-MS experimental process:
Experimental Steps:
- HDX Labeling Initiation: Dilute the refolding reaction (chaperone-client complex) into deuterated buffer (e.g., 10 mM potassium phosphate in D₂O, pD 6.6-7.0). Maintain constant temperature (typically 20-25°C) throughout labeling [81] [82].
- Labeling Time Course: Aliquot samples at multiple time points (e.g., 10 s, 100 s, 1000 s) to capture both fast and slow exchanging amides [82].
- Quenching: For each time point, transfer aliquot to low pH (pH 2.5) and low temperature (0°C) conditions (e.g., equal volume of quench buffer: 2% formic acid). This reduces the HDX rate approximately 10,000-fold [81].
- Proteolytic Digestion: Pass quenched samples through an immobilized pepsin column (20°C) for rapid digestion (approximately 1-3 minutes). In some cases, adding reducing agents like tris(2-carboxyethyl)phosphine hydrochloride may be necessary for proteins with disulfide bonds [81] [82].
- Liquid Chromatography: Trap and desalt peptides on a C18 pre-column at 0.5°C to minimize back-exchange, then separate using a reverse-phase C18 analytical column with a gradient of acetonitrile (0.1% formic acid) [81].
- Mass Spectrometry Analysis: Analyze peptides using high-resolution mass spectrometry (e.g., Synapt G2Si HDMS). Data acquisition in MSE mode with ion mobility separation enhances peptide identification [81].
- Data Processing: Identify peptides and measure deuterium incorporation using specialized software (e.g., DynamX, HDExaminer). Correct for back-exchange using fully deuterated controls, and express results as relative fractional uptake [81].
Key Research Findings and Data Interpretation
HDX-MS studies have yielded fundamental insights into chaperone mechanisms and protein folding pathways. Recent research on cotranslational folding of E. coli dihydrofolate reductase (DHFR) revealed that the ribosome allows nascent chains to access structured intermediates not observed during refolding from denaturant. These intermediates form while the C-terminal sequences remain confined in the ribosome exit tunnel, with Trigger Factor binding partially folded states without disrupting their structure [82].
Proteome-wide studies of chaperone-assisted refolding in E. coli have demonstrated that GroEL can refold approximately 85% of detectable proteins, showing particular importance for acidic proteins and those with high molecular weight. This refolding capability overlaps significantly with the DnaK (Hsp70) system, suggesting a common mechanism of unfolding misfolded states despite their vastly different structures [64].
The table below summarizes key quantitative findings from recent HDX-MS studies of chaperone-assisted folding:
Table 1: Key Quantitative Findings from HDX-MS Studies of Chaperone-Assisted Folding
| Chaperone System | Experimental System | Key Finding | Reference |
|---|---|---|---|
| GroEL/ES | E. coli proteome | Refolds ~85% of detectable proteins, especially acidic (pI) and high molecular weight proteins | [64] |
| Trigger Factor | Ribosome-nascent chain complexes (1-106RNC) | Binds partially folded states without disrupting intermediate structure | [82] |
| Hsp70 | SH3 client protein folding | Closed-lid state interacts with client via conserved nonpolar residues, preventing nonnative hydrophobic collapse | [6] |
| Ribosome-associated folding | DHFR nascent chains | Folding pathway distinct from denaturant-refolding; DLD subdomain folding delayed until C-terminus released | [82] |
Spectroscopic Techniques for Monitoring Refolding
Limited Proteolysis-Mass Spectrometry (LiP-MS)
Limited proteolysis coupled with mass spectrometry (LiP-MS) has emerged as a powerful spectroscopic method for globally monitoring protein structures during refolding. This technique leverages the sensitivity of proteolytic cleavage to local protein flexibility and solvent accessibility, providing structural information across entire proteomes [64].
The LiP-MS protocol for refolding analysis involves unfolding proteins in 6 M guanidinium chloride, followed by rapid dilution into native conditions. Pulse proteolysis with proteinase K (PK) then cleaves regions that remain solvent-exposed or flexible. LC-MS/MS identifies and quantifies the resulting peptide fragments, comparing their abundances to native controls that were never unfolded. Regions showing differential proteolytic susceptibility indicate failure to regain native structure [64].
Application of LiP-MS to E. coli proteome refolding revealed that protein isoelectric point (pI) strongly influences refoldability, with basic proteins generally refolding efficiently in cytosolic milieu, while acidic proteins show greater dependency on GroEL assistance. The technique has also identified specific protein cohorts that resist refolding even with chaperone assistance, suggesting adaptation to cotranslational folding [64].
Fluorescence and Circular Dichroism Spectroscopy
While not explicitly detailed in the search results, fluorescence spectroscopy (particularly intrinsic tryptophan fluorescence) and circular dichroism (CD) are widely used in protein refolding studies. Fluorescence spectroscopy monitors changes in the local environment of aromatic residues as folding proceeds, while CD provides information about secondary structure content (far-UV) and tertiary structure (near-UV).
These techniques are particularly valuable for monitoring refolding kinetics, as they can track structural changes in real-time with high temporal resolution. When combined with HDX-MS, they provide complementary information about both global structural features and local conformational dynamics during chaperone-assisted refolding.
Chromatographic Methods in Refolding Studies
Analytical Size Exclusion Chromatography (SEC)
Size exclusion chromatography serves as a critical tool for assessing the oligomeric state and aggregation status of refolding proteins. By separating molecules based on their hydrodynamic radius, SEC can distinguish between properly folded monomers, folding intermediates, and aggregated species that often form during refolding processes [83] [22].
In refolding experiments, SEC is particularly valuable for:
- Quantifying the distribution between soluble and aggregated species
- Monitoring the time-dependent progression from unfolded to folded states
- Assessing the effectiveness of chaperones in preventing aggregation
- Identifying stable folding intermediates based on their elution volumes
The application of SEC to chaperone-assisted refolding has demonstrated that molecular chaperones significantly reduce the formation of high-molecular-weight aggregates while promoting the accumulation of monomeric, properly folded proteins [83].
High-Performance Liquid Chromatography (HPLC)
Reverse-phase HPLC provides complementary information to SEC by separating proteins and peptides based on hydrophobicity. In refolding studies, HPLC is particularly useful for monitoring disulfide bond formation and analyzing proteolytic fragments generated in LiP-MS experiments [38].
Recent advances have focused on developing HPLC methodologies capable of quantifying different folding states during refolding processes. These approaches enable researchers to track the conversion of folding intermediates (I) to native (N) states while competing against aggregation (A) pathways. The sensitivity of HPLC makes it particularly valuable for analyzing refolding processes at low protein concentrations (typically <1 g/L), which are common in refolding experiments aimed at minimizing aggregation [38].
Table 2: Chromatographic and Spectroscopic Methods for Refolding Analysis
| Method | Key Measurement | Information Provided | Typical Sample Requirements |
|---|---|---|---|
| Analytical SEC | Hydrodynamic radius | Oligomeric state, aggregation status | 0.1-1 mg/mL, 50-100 μL injection |
| Reverse-Phase HPLC | Hydrophobicity | Disulfide bond status, peptide mapping | Low μg range, <1 mg/mL |
| LiP-MS | Proteolytic susceptibility | Global structural states, flexible regions | Complex mixtures, proteome-wide |
| Fluorescence Spectroscopy | Tryptophan environment | Tertiary structure formation, kinetics | ~0.1-1 mg/mL, 100-500 μL |
| Circular Dichroism | Secondary structure | α-helix/β-sheet content, folding stability | ~0.1-0.5 mg/mL, >200 μL |
Integrated Experimental Design for Chaperone Studies
Workflow for Comprehensive Folding Analysis
A comprehensive analysis of chaperone-assisted refolding requires integrating multiple biophysical techniques to capture different aspects of the folding process. The following workflow illustrates how these methods can be combined in a typical experimental design:
Research Reagent Solutions
The following table outlines essential reagents and materials for biophysical studies of chaperone-assisted refolding:
Table 3: Essential Research Reagents for Chaperone-Assisted Refolding Studies
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| Molecular Chaperones | Refolding assistance | GroEL/ES, DnaK/DnaJ/GrpE (Hsp70/40), Trigger Factor |
| Deuterium Oxide (D₂O) | HDX-MS labeling | 99.9% deuterium purity, buffered to appropriate pD |
| Chaotropic Agents | Denaturation and solubilization | Urea (ultrapure), Guanidine HCl (sequanal grade) |
| Proteases | Digestion for HDX-MS and LiP-MS | Immobilized pepsin, Proteinase K (for LiP-MS) |
| Chromatography Columns | Separation and analysis | SEC columns (e.g., Superdex), RP-HPLC (C18), immobilized pepsin |
| Refolding Additives | Enhancing refolding yields | L-arginine, glycerol, detergents, redox shufflers |
| Mass Spectrometry Standards | Calibration and quantification | Leu-enkephalin, sodium iodide |
The integration of HDX-MS, spectroscopic methods, and chromatographic techniques provides a powerful multidimensional approach for studying chaperone-assisted protein refolding. HDX-MS excels at mapping local conformational dynamics and binding interfaces with peptide-level resolution. LiP-MS offers a global perspective on structural states across complex protein mixtures, while SEC and HPLC monitor oligomeric states and aggregation behavior. Together, these methods have revealed fundamental principles of chaperone mechanisms, including the distinct folding pathways accessed during cotranslational folding, the client specificity of different chaperone systems, and the key structural features that determine protein refoldability.
As these biophysical techniques continue to advance in sensitivity and throughput, they will enable increasingly detailed understanding of how chaperones navigate the energy landscapes of their client proteins. This knowledge is essential for developing improved protein refolding protocols for biotechnology and therapeutic applications, and for understanding the fundamental principles governing protein homeostasis in living cells.
Molecular chaperones are essential components of the cellular proteostasis network, preventing protein aggregation and facilitating the correct folding of substrate proteins into their functional, native states [61]. A fundamental challenge in structural biology has been visualizing the highly dynamic and transient interactions between chaperones and their folding substrates [63]. Conventional structural techniques like X-ray crystallography often struggle to resolve such heterogeneous complexes, as disordered or multi-conformational regions typically yield fragmented electron density that is difficult to interpret using traditional methods [63].
The Residual Electron and Anomalous Density (READ) method represents a significant methodological advancement that addresses this challenge. Developed to visualize the ensemble of conformations that a substrate protein samples while bound to its chaperone, READ combines complementary structural techniques to resolve dynamic complexes at high resolution [63]. This approach has provided unprecedented insights into the mechanism of chaperone-assisted protein folding, revealing how substrates can explore their folding landscape while associated with chaperones like the E. coli chaperone Spy [63].
This protocol details the application of the READ method, framing it within the broader context of researching chaperone-assisted protein refolding mechanisms. The technique is particularly valuable for researchers investigating how chaperones guide the folding of intrinsically disordered proteins, conditionally folded intermediates, and other dynamic substrates that have eluded conventional structural analysis.
Background
The Challenge of Structural Heterogeneity
Traditional X-ray crystallography typically relies on biomolecules adopting nearly identical conformations throughout the crystal lattice. However, chaperone-substrate complexes are inherently dynamic systems where the substrate may exist in an ensemble of conformations ranging from unfolded to partially folded and native-like states [63]. In standard crystallographic analysis, this heterogeneity manifests as discontinuous, fragmented electron density for the substrate region, which is often discarded as uninterpretable, thereby losing crucial information about the folding process [63].
Spy-Im7: A Model Chaperone-Substrate System
The READ method was developed using the E. coli chaperone Spy and its substrate protein Im7 (immunity protein 7) [63]. Spy is an ATP-independent periplasmic chaperone that forms a thin α-helical homodimeric cradle. It prevents protein aggregation and aids in protein folding under various stress conditions [63]. The Im7 protein, specifically a fragment encompassing residues 6-45 (Im7(_{6-45})), which constitutes the entire Spy-binding region, served as an ideal model substrate because NMR data indicated it could recapitulate unfolded, partially folded, and native-like states of Im7 [63].
READ Method Workflow
The READ method integrates multiple experimental and computational approaches in a five-step process, outlined schematically below and detailed in the subsequent sections.
Step 1: Crystallization and Initial Structure Determination
Protocol
- Complex Crystallization: Co-crystallize the chaperone (e.g., Spy) with its substrate protein (e.g., Im7(_{6-45})) using screening approaches that yield crystals only in the presence of the substrate. This suggests the substrate is incorporated and essential for crystal formation [63].
- Data Collection: Collect high-resolution X-ray diffraction data from obtained co-crystals. For the Spy-Im7 system, crystals typically diffracted to ~1.8 Å resolution [63].
- Initial Phasing: Solve the structure of the well-ordered chaperone portion using standard crystallographic methods (e.g., SAD with selenomethionine-labeled Spy) [63].
- Residual Density Identification: Examine the resulting electron density maps for fragmented, discontinuous density in the substrate-binding region, which indicates conformational heterogeneity of the substrate.
Step 2: Incorporation of Anomalous Scatterers
Protocol
- Residue Selection: Identify multiple solvent-accessible residues throughout the substrate sequence for anomalous scatterer incorporation. In the Im7(_{6-45}) study, eight residues were selected [63].
- Amino Acid Substitution: Individually substitute each selected residue with the non-canonical amino acid 4-iodophenylalanine (pI-Phe), which contains iodine—a strong anomalous scatterer [63]. This generates a series of substrate variants, each with a single pI-Phe substitution at a distinct position.
- Complex Preparation and Crystallization: Co-crystallize the chaperone with each individual pI-Phe-labeled substrate variant under identical conditions used for the wild-type complex.
- Anomalous Data Collection: For each variant crystal, collect X-ray diffraction data and anomalous diffraction data to locate the iodine atoms in three-dimensional space.
Table 1: Essential Research Reagents for READ Method Implementation
| Reagent | Function/Description | Application in READ |
|---|---|---|
| Chaperone Protein | Well-characterized chaperone (e.g., Spy) | Forms the static component of the complex; provides initial phasing |
| Substrate Protein | Dynamic client protein (e.g., Im7(_{6-45})) | Target for conformational ensemble analysis |
| 4-Iodophenylalanine (pI-Phe) | Non-canonical amino acid with strong anomalous scatterer (iodine) | Site-specific labeling of substrate; provides spatial landmarks via anomalous signals |
| Crystallization Reagents | Standard screening and optimization kits | Production of well-diffracting chaperone-substrate co-crystals |
| Molecular Dynamics Software | Simulation packages (e.g., GROMACS, AMBER) | Generation of diverse conformational pool for sample-and-select procedure |
Step 3: Molecular Dynamics Simulations
Protocol
- System Setup: Construct initial models of the chaperone-substrate complex with the substrate in an extended conformation.
- Coarse-Grained Simulations: Perform molecular dynamics simulations using a coarse-grained, single-residue resolution model for protein folding and chaperone-substrate binding [63]. These simulations should allow the substrate to repeatedly bind and unbind from the chaperone to sample a wide conformational space.
- Conformational Sampling: Extract a diverse pool of chaperone-substrate complexes (approximately 10,000 structures) from the trajectories to be used in the ensemble selection process [63].
Step 4: Sample-and-Select Ensemble Determination
Protocol
- Data Preparation: Prepare compressed versions of both the residual electron density map (2mFo−DFc) and the anomalous difference maps for computational efficiency during the selection process [63].
- Genetic Algorithm Implementation: Employ a genetic algorithm that iteratively constructs and evaluates structural ensembles against the experimental data [63]. The algorithm:
- Randomly selects a small number of conformations (e.g., 5-10 structures) from the MD-generated pool.
- Calculates how well the selected ensemble reproduces both the residual electron density and the anomalous signals from all pI-Phe labels.
- Iteratively modifies the ensemble composition through genetic algorithm operations (mutation, crossover, selection).
- Optimization Criterion: Identify the minimal ensemble of conformations that collectively best satisfies both the residual electron density and the anomalous scattering data [63].
Step 5: Validation
Protocol
- Cross-Validation: Use standard crystallographic cross-validation methods (e.g., R(_{free})) to ensure the ensemble model does not overfit the data [63].
- Statistical Analysis: Apply multiple statistical tests to validate the ensemble against the experimental data, ensuring the selected conformations provide a physically realistic representation of the substrate's conformational landscape [63].
Data Analysis and Interpretation
Quantitative Metrics
The READ method generates both structural and quantitative data on the conformational ensemble of the chaperone-bound substrate. Key analytical approaches include:
Table 2: Representative Anomalous Scattering Data from Spy-Im7 READ Analysis
| pI-Phe Position | Number of Anomalous Sites Detected | Relative Occupancy Patterns | Structural Interpretation |
|---|---|---|---|
| Residue 10 | 3 | High/Medium/Low | Substrate samples multiple distinct regions of chaperone cradle |
| Residue 15 | 2 | High/Medium | Partial conformational preference |
| Residue 22 | 4 | High/Medium/Medium/Low | Highly flexible region with diverse binding modes |
| Residue 31 | 3 | High/Medium/Low | Distributed binding conformations |
| Residue 38 | 2 | High/Low | Bimodal conformational sampling |
Structural Interpretation
The final output of the READ analysis is a structural ensemble depicting the various folding states of the substrate while bound to the chaperone. In the Spy-Im7 system, this ensemble revealed that Im7 samples conformations ranging from unfolded to partially folded and native-like states while associated with Spy [63]. This demonstrated that substrates can explore their folding landscape while bound to a chaperone, rather than being locked in a single conformation.
The diagram below illustrates the conformational landscape accessible to a substrate while chaperone-bound, as revealed by READ analysis.
Applications in Chaperone-Assisted Refolding Research
The READ method provides unique insights into fundamental questions in protein folding:
- Folding Pathways: Reveals how chaperones allow substrates to sample native-like conformations while bound, providing direct structural evidence for folding during chaperone association [63].
- Chaperone Mechanisms: Distinguishes between passive "holdase" versus active "foldase" chaperone mechanisms by visualizing whether substrates remain static or dynamically sample folding states while chaperone-bound.
- Aggregation Prevention: Illustrates how chaperones like Spy provide a protected environment where substrates can explore conformational space without engaging in off-pathway aggregation [63].
- Energy Landscape Theory: Provides experimental validation for the folding funnel concept by showing that chaperone-bound substrates can access multiple folding intermediates on the path to the native state [63] [61].
Technical Considerations and Limitations
Advantages
- Heterogeneity Resolution: Uniquely capable of resolving multiple coexisting conformations in crystalline materials.
- Complementary Data: Combines the strengths of crystallography (high resolution) and NMR (ensemble information).
- Site-Specific Information: Anomalous scattering provides precise spatial constraints for labeled residues.
- General Applicability: The approach can be adapted to other dynamic protein complexes beyond chaperone-substrate systems.
Challenges
- Technical Complexity: Requires expertise in multiple techniques including protein engineering, crystallography, and computational methods.
- Resource Intensive: Numerous crystals must be prepared and analyzed for comprehensive anomalous scattering data.
- Labeling Considerations: pI-Phe incorporation must not significantly alter substrate binding or chaperone function.
- Computational Demand: MD simulations and ensemble selection algorithms require significant computational resources.
The READ method represents a significant advance in structural biology, providing a powerful approach to visualize dynamic chaperone-substrate complexes that have previously eluded high-resolution analysis. By integrating residual electron density, anomalous scattering, and computational modeling, READ enables researchers to obtain structural ensembles that reveal how chaperones interact with folding substrates at unprecedented resolution.
This protocol outlines the comprehensive application of the READ method, from initial crystallization through final ensemble validation. The technique is particularly valuable for research on chaperone-assisted protein refolding, as it provides direct structural insights into the molecular mechanisms by which chaperones guide protein folding, prevent aggregation, and maintain proteostasis. As structural biology continues to confront increasingly dynamic biological systems, approaches like READ will be essential for revealing the structural basis of complex biomolecular processes.
Within the complex cellular environment, newly synthesized polypeptides face the fundamental challenge of navigating the energy landscape to attain their native, functional three-dimensional structures. This folding process occurs through two principal mechanisms: cotranslational folding, which proceeds as the polypeptide chain emerges sequentially from the ribosome, and post-translational folding, which occurs after the complete protein has been released into the cellular milieu. Understanding the distinctions between these pathways is not merely academic; it forms the critical foundation for developing effective chaperone-assisted refolding protocols for research and therapeutic applications. Misfolded proteins not only lose functionality but can also aggregate, contributing to various neurodegenerative diseases and other proteinopathies. This analysis provides a structured comparison of these folding pathways, supported by quantitative data and experimental methodologies, to inform the design of targeted refolding strategies in both basic research and drug development contexts.
Fundamental Mechanisms and Key Distinctions
Cotranslational Folding
Cotranslational folding occurs vectorially as the nascent polypeptide chain is synthesized and extruded through the ribosomal exit tunnel (RET). This tunnel imposes physical constraints that profoundly influence the folding process. The confined space within the RET restricts long-range contacts and promotes the formation of α-helical secondary structures due to its dimensions and chemical properties [84]. As the chain emerges, folding domains can begin to organize sequentially, often starting with the N-terminal domains. The translation speed, influenced by factors like codon usage, provides a tunable cellular mechanism to regulate folding kinetics, with slower rates allowing more time for structured intermediates to form [84]. The ribosome itself acts as a passive chaperone, with studies on HaloTag demonstrating that it can reroute folding pathways to bypass aggregation-prone intermediates observed during refolding, thereby increasing folding efficiency [85].
Post-Translational Folding
In contrast, post-translational folding involves a full-length, newly synthesized polypeptide that must navigate its entire energy landscape without sequential constraints. This process often relies heavily on molecular chaperones such as the Hsp70 system (DnaK/DnaJ/GrpE in E. coli) and the chaperonin system (GroEL/ES in E. coli) to prevent aggregation and facilitate proper folding [64]. These chaperones recognize exposed hydrophobic patches on non-native structures, providing a protected environment for folding to proceed. The "chaperone code" – a combinatorial array of post-translational modifications (PTMs) on chaperones including phosphorylation, acetylation, and ubiquitination – fine-tunes their activity, specificity, and subcellular localization, adding a critical regulatory layer to the folding process [86] [87]. For instance, phosphorylation of Hsp70 at tyrosine 525 has been shown to increase its nuclear accumulation and promote cell survival following heat shock [86].
Table 1: Core Characteristics of Folding Pathways
| Feature | Cotranslational Folding | Post-Translational Folding |
|---|---|---|
| Spatial Context | Ribosomal Exit Tunnel (RET) and emerging chain | Cytosol, ER, or other cellular compartments |
| Key Influences | RET geometry, translation speed, nascent chain sequence | Chaperone availability, "Chaperone Code" (PTMs), cellular environment |
| Structural Outcome | More helix-rich, fewer non-native long-range contacts | Dependent on intrinsic landscape and chaperone assistance |
| Primary Chaperone System | Ribosome as passive chaperone | Hsp70/DnaK and Hsp60/GroEL systems |
| Regulatory Mechanism | Codon usage/translation speed | Post-translational modifications of chaperones |
Quantitative Comparison of Folding Outcomes
Recent computational and experimental studies have provided quantitative insights into how these folding pathways differ in their structural outcomes and efficiency.
Structural Properties of Nascent Chains
Extensive molecular dynamics simulations comparing cotranslational and free folding (an in vitro model for post-translational folding) have revealed significant differences in the initial structural ensembles. The data demonstrate that upon expulsion from the RET, the nascent peptide adopts a configuration with a significantly higher helical population compared to structures formed during free folding. For instance, the average helical population (APHelix) for proteins like NTL9 was markedly higher across all tested translation speeds in cotranslational folding scenarios [84]. Conversely, the formation of β-sheets was generally suppressed during cotranslational folding, with the average population of residues forming β-sheets (APSheet) being significantly lower than in free folding. Furthermore, cotranslational folding reduces long-range contacts—both native (APnat) and nonnative (APnon)—in the emerging peptide, guiding it toward a less entangled starting configuration for subsequent folding [84].
Chaperone Dependence and Proteome-Wide Refolding Efficiency
A proteome-wide refolding assay in E. coli using limited proteolysis-mass spectrometry (LiP-MS) has systematically quantified chaperone requirements, highlighting a fundamental relationship between protein properties and folding pathway dependence. The study found that GroEL/ES can refold the majority (85%) of the E. coli proteins for which data was obtained. Notably, dependency was not random; acidic proteins (low pI) and proteins with high molecular weight were particularly reliant on GroEL assistance. These proteins likely present folding challenges—such as increased surface charge repulsion or complex domain integration—that the chaperonin system is uniquely equipped to handle. The Hsp70 system (DnaK/DnaJ/GrpE) was shown to refold a largely overlapping set of proteins, suggesting a common mechanism of resolving misfolded states despite their vastly different structures [64]. Finally, a cohort of proteins was identified as intransigent to refolding by either chaperone system, suggesting these may be specialized for efficient cotranslational folding, after which they remain kinetically trapped in their native states [64].
Table 2: Quantitative Comparison of Folding Outcomes
| Parameter | Cotranslational Folding | Post-Translational Folding | Experimental/Simulation Basis |
|---|---|---|---|
| Helical Population (APHelix) | Significantly higher | Lower | MD simulations (e.g., NTL9) [84] |
| β-Sheet Population (APSheet) | Significantly lower | Higher | MD simulations (GTT, NTL9, HOME) [84] |
| Long-Range Native Contacts | Reduced (except at fastest speeds) | More prevalent | MD simulations, contact distribution analysis [84] |
| Chaperone Dependence (GroEL) | Not applicable | 85% of E. coli proteome refoldable | LiP-MS proteome-wide refolding assay [64] |
| Key Client Features for Chaperones | N/A | Acidic pI, High MW, Multi-domain | Proteomic analysis of refoldability [64] |
Experimental Protocols and Methodologies
Protocol A: Computational Simulation of Cotranslational Folding
The General Protein Cotranslational Folding (GPCTF) simulation framework provides a methodology for studying cotranslational folding in silico [84] [88].
- System Setup: Model the ribosomal exit tunnel (RET) as a rigid, geometrically constrained tunnel. The translation process is simulated by incrementally shifting this tunnel model along the nascent chain at predefined time intervals (e.g., every 100 ns per residue).
- Simulation Execution:
- Residues within the tunnel are permitted to fold and undergo fully elastic collisions with the tunnel wall.
- Residues outside the tunnel are restricted by an oscillator potential and do not participate in folding.
- Once all residues have entered the tunnel, the model is held stationary, allowing the peptide to diffuse out.
- After complete exit, the tunnel model is removed, and the peptide continues folding freely.
- Data Collection and Analysis:
- Secondary Structure Analysis: Calculate the population/probability of each residue forming a helix (or β-sheet) in the structural ensemble upon exit.
- Contact Analysis: Calculate the population of native (APnat) and nonnative (APnon) contacts. Compare contact distributions, focusing on short-range vs. long-range interactions.
- Pathway Clustering: After defining the folded (Q > 0.8) and unfolded (Q < 0.2) states based on the fraction of native contacts (Q-value), identified folding pathways are clustered and analyzed based on Φ-value analysis.
Protocol B: Proteome-Wide Assessment of Refolding and Chaperone Dependence
This protocol uses limited proteolysis-mass spectrometry (LiP-MS) to globally monitor refolding outcomes in a complex mixture, such as a cell extract [64].
- Sample Preparation:
- Prepare E. coli lysates in a native-like cytosolic medium ("cyto-serum").
- Denaturation: Fully unfold the proteome by overnight incubation in 6 M Guanidinium Chloride (GdmCl).
- Refolding Initiation: Rapidly dilute the denatured mixture into native conditions. Test conditions include: (a) cytosolic medium alone, (b) with added GroEL/ES (Hsp60), and (c) with added DnaK/DnaJ/GrpE (Hsp70/40).
- Structural Probing via Limited Proteolysis:
- Subject the refolding reactions to pulse proteolysis with Proteinase K (PK). PK cleaves peptide bonds in solvent-exposed or flexible regions, serving as a probe for unstructured or misfolded regions.
- Quench the proteolysis reaction.
- Mass Spectrometry Analysis and Data Processing:
- Digest the samples with a standard protease (e.g., trypsin) and analyze using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS).
- Sequence and quantify tens of thousands of peptide fragments.
- Compare the limited proteolysis profiles of the refolded samples against a "native" control (a sample never exposed to denaturant).
- Proteins displaying significantly different proteolytic patterns in the refolded vs. native sample are classified as "non-refoldable" under the given conditions. Proteins whose patterns normalize in the presence of a chaperone are classified as dependent on that chaperone for refolding.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Protein Folding Research
| Reagent / Material | Primary Function | Application Context |
|---|---|---|
| GroEL/ES (Hsp60) | Chaperonin system; provides a central folding chamber for individual proteins. | Refolding assays for obligate clients, particularly acidic, high MW, and α/β proteins [64]. |
| DnaK/DnaJ/GrpE (Hsp70) | Hsp70 system; binds hydrophobic patches, prevents aggregation, promotes (re)folding. | Broad-spectrum refolding assays; studying early folding intermediates; disaggregation [64]. |
| Guanidinium Chloride (GdmCl) | Chemical denaturant; unfolds proteins by disrupting non-covalent interactions. | Preparation of unfolded starting material for refolding assays (e.g., at 6 M concentration) [64]. |
| Proteinase K | Broad-spectrum serine protease; cleaves at solvent-exposed/flexible regions. | Limited Proteolysis (LiP) to probe protein structure and identify misfolded states [64]. |
| GPCTF Framework | Computational model simulating the ribosomal exit tunnel and translation. | In silico studies of cotranslational folding kinetics and pathway regulation [84] [88]. |
| AlphaFold DB & Foldseek | Protein structure database and fast structure search tool. | Source of evolutionary data for designing folding force fields (e.g., in FoldPAthreader) [89]. |
Application Notes for Chaperone-Assisted Refolding Protocols
The comparative analysis of folding pathways directly translates into practical strategies for refolding proteins in research and bioprocessing.
Prioritize Chaperone Systems Based on Protein Characteristics: When designing a refolding protocol for a poorly characterized protein, use the client profiles from proteomic studies as a guide. For acidic proteins (low pI) and large, multi-domain proteins, prioritize the use of the GroEL/ES system first, as these are its hallmark clients. For a broader range of proteins, particularly those prone to aggregation, the DnaK (Hsp70) system is an excellent starting point. Consider using both systems in tandem, as they can operate cooperatively [64].
Leverage Cotranslational Folding Principles for Heterologous Expression: Optimize the heterologous expression of difficult-to-fold proteins by manipulating translation kinetics. This can be achieved by engineering the gene sequence to use rarer codons at critical points where slowing translation might allow proper domain folding or by introducing synonymous codon substitutions to modulate the translation speed without altering the amino acid sequence [84].
Harness Evolutionary Information for Folding Prediction: For targets where experimental folding data is scarce, use computational tools like FoldPAthreader. These tools leverage evolutionary information from massive structure databases (e.g., AlphaFold DB) to predict folding pathways and intermediates, providing a rational basis for designing refolding experiments and interpreting their results [89].
Account for the Regulatory "Chaperone Code": In cellular or complex in vitro refolding assays, remember that chaperone activity is dynamically regulated by PTMs. The efficiency of a refolding reaction can be influenced by the phosphorylation, acetylation, or methylation status of the chaperones themselves. Incorporating relevant kinase or phosphatase activities (or their inhibitors) into the refolding buffer may be necessary to modulate chaperone function optimally [86] [87].
The chaperone system (CS), a sophisticated network comprising molecular chaperones, co-chaperones, and chaperone receptors, serves as the fundamental guardian of cellular proteostasis by regulating protein folding, quality control, and degradation [90] [33]. At the core of this system are the highly conserved heat shock proteins (HSPs), which are categorized by molecular weight into families such as HSP100, HSP90, HSP70, HSP60, HSP40, and small HSPs (sHSPs) [33]. Under physiological conditions, these molecular chaperones prevent protein misfolding and aggregation, but their dysregulation represents a critical pathogenic mechanism in diverse human diseases [90] [91]. In cancer, numerous HSPs are abnormally overexpressed, where they stabilize oncoproteins, promote tumor growth, and confer resistance to therapy [90] [33]. Conversely, in neurodegenerative disorders such as Alzheimer's and Parkinson's disease, chaperone dysfunction contributes to the accumulation of toxic protein aggregates that characterize these conditions [91] [92]. This dichotomy presents two therapeutic avenues: "negative chaperonotherapy," which inhibits pro-disease chaperones, and "positive chaperonotherapy," which enhances or restores protective chaperone functions [90]. These approaches are now yielding promising clinical strategies for some of the most challenging human diseases.
Chaperone Dysregulation in Disease Pathogenesis
Cancer: Hijacking the Proteostasis Network
Cancer cells exploit the chaperone system to support their rapid proliferation, evade apoptosis, and develop therapeutic resistance. In aggressive nervous system tumors like glioblastoma (GBM) and neuroblastoma (NB), multiple HSPs are constitutively overexpressed and correlate strongly with poor prognosis [90]. These chaperones stabilize oncoproteins, activate pro-survival signaling pathways, and protect against proteotoxic stress in the hostile tumor microenvironment characterized by hypoxia, acidosis, and oxidative stress [90]. HSP90, for instance, supports over 300 client proteins, many of which are oncoproteins, transcription factors, and critical signaling nodes [93]. The malignant reorganization of the chaperone network extends to the formation of "epichaperome" complexes—stable, high-molecular-weight structures that emerge in over 50% of malignancies and enhance the proteostatic capacity of cancer cells under oncogenic stress [93].
Neurodegeneration: Collapse of Protein Quality Control
Neurodegenerative diseases share a common mechanism in the accumulation of misfolded proteins that aggregate and become toxic to neurons [91]. In Alzheimer's disease (AD), the pathological hallmarks include extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau [92]. The aging brain experiences a progressive decline in proteostasis network functionality, with reduced chaperone activity and impaired protein degradation systems creating an environment permissive for protein aggregation [3]. Small HSPs like HSP27, which serve as the first line of defense against protein misfolding, become overwhelmed, leading to the irreversible aggregation of amyloidogenic proteins [33]. Recent research has highlighted the endoplasmic reticulum (ER) resident chaperone GRP78 (HSPA5) as a key modulator in AD, where its dysfunction exacerbates ER stress and contributes to the pathological accumulation of misfolded proteins [90] [92].
Therapeutic Targeting Strategies
Negative Chaperonotherapy: Inhibiting Pro-Disease Chaperones
Negative chaperonotherapy aims to inhibit pro-tumorigenic or otherwise pathogenic chaperones. This approach has shown significant promise in oncology, particularly for aggressive cancers like GBM and NB [90].
Table 1: Selected Negative Chaperonotherapy Agents in Preclinical and Clinical Development
| Target | Therapeutic Agent | Disease Context | Mechanism of Action | Development Stage |
|---|---|---|---|---|
| HSP90 | 17-AAG (Tanespimycin) | Glioma, Neuroblastoma | Inhibits HSP90 ATPase activity; degrades oncogenic client proteins (Raf-1, AKT) | Preclinical models [90] |
| HSP90 | NXD30001 | Glioblastoma | Targets EGFR-PI3K-AKT axis; increases radiosensitivity | Preclinical models [90] |
| HSP90 | PU-H71 | Various Cancers | Preferentially binds HSP90 in epichaperome complexes | Clinical Trials [93] |
| HSP70 | Pifithrin-μ | Glioblastoma | Inhibits HSP70; activates pro-apoptotic UPR cascades | Preclinical models [90] |
| HSPA5/GRP78 | OSU-03012 | Glioblastoma | Induces ER stress; suppresses tumor growth and enhances radiation | Preclinical models [90] |
| HSP27 | NA49 | Various Cancers | Cross-links HSP27 proteins to prevent functional oligomerization | Preclinical [93] |
The clinical translation of HSP90 inhibitors illustrates both the promise and challenges of this approach. While inhibitors like TAS116 (pimitespib) have gained approval, others have faced limitations including limited therapeutic efficacy and toxicity [93]. Emerging strategies now focus on disrupting specific protein-protein interactions (PPIs) within the chaperone network rather than general inhibition of chaperone activity, potentially offering enhanced selectivity and reduced toxicity [33] [93].
Positive Chaperonotherapy and Immunomodulation
Positive chaperonotherapy aims to enhance or restore the beneficial functions of chaperones. In neurodegenerative diseases, this strategy seeks to boost the cellular defense systems that prevent protein aggregation. A striking example is the recent discovery that the common amino acid arginine acts as a safe chemical chaperone that can markedly reduce Aβ aggregation and its toxic effects in animal models of AD [94]. Oral arginine administration in Drosophila and mouse models lowered plaque levels, reduced neuroinflammation, and improved behavioral outcomes, presenting a promising, low-cost therapeutic candidate for repurposing [94].
In oncology, extracellular chaperones can stimulate anti-tumor immunity. Glioma cells exposed to stress constitutively express HSP70 on their plasma membrane, where it functions as a danger signal that primes natural killer (NK) and dendritic cells [90]. Clinical correlative studies show that glioblastoma patients with elevated frequencies of CD56+/CD94+/CD69+ NK cells, which recognize membrane-HSP70, have significantly longer overall survival [90]. This provides the rationale for chaperone-based immunotherapies that leverage the immunomodulatory properties of extracellular HSPs.
Application Notes & Experimental Protocols
Protocol 1: Evaluating HSP90 Inhibitor Efficacy in Glioblastoma Models
Background: This protocol details the assessment of HSP90 inhibitor efficacy using in vitro and in vivo glioblastoma models, based on methodologies successfully employed in preclinical studies [90].
Materials:
- Patient-derived or immortalized GBM cell lines
- HSP90 inhibitor (e.g., 17-AAG, NXD30001, AUY922)
- Orthotopic glioma mouse model
- Cell viability assay kit (e.g., MTT, CellTiter-Glo)
- Apoptosis detection kit (Annexin V/PI)
- Western blot equipment and antibodies (HSP90 client proteins: AKT, Cdk4; apoptosis markers: cleaved PARP, caspase-3)
Procedure:
- In Vitro Dose-Response Analysis:
- Seed GBM cells in 96-well plates (3,000-5,000 cells/well).
- After 24 hours, treat with HSP90 inhibitor across a concentration gradient (e.g., 1 nM - 100 μM) for 72 hours.
- Assess cell viability using MTT or CellTiter-Glo assay according to manufacturer instructions.
- Calculate IC50 values using non-linear regression analysis.
Mechanistic Validation via Western Blot:
- Treat GBM cells with IC50 concentration of HSP90 inhibitor for 24-48 hours.
- Lyse cells and quantify protein concentration.
- Perform Western blotting to detect reduction in client proteins (AKT, Cdk4) and induction of apoptosis markers (cleaved PARP).
In Vivo Efficacy Study:
- Implant GBM cells intracranially into immunocompromised mice.
- Once tumors are established (confirmed by imaging), randomize mice into treatment and control groups.
- Administer HSP90 inhibitor or vehicle control via predetermined route (e.g., intraperitoneal injection) for 4-6 weeks.
- Monitor tumor growth weekly via bioluminescence imaging or MRI.
- Evaluate survival benefit using Kaplan-Meier analysis.
Expected Outcomes: Effective HSP90 inhibitors should significantly reduce client protein levels, induce apoptosis in vitro, and inhibit tumor growth while extending survival in orthotopic models [90].
Protocol 2: Assessing Chemical Chaperones in Alzheimer's Models
Background: This protocol describes the evaluation of chemical chaperones (e.g., arginine) for suppressing Aβ aggregation using in vitro and in vivo Alzheimer's disease models [94].
Materials:
- Synthetic Aβ42 peptide
- L-arginine
- Aβ aggregation assay kit (Thioflavin T fluorescence-based)
- AppNL-G-F knock-in mouse model or Aβ42-expressing Drosophila model
- Behavioral testing apparatus (e.g., Morris water maze, Y-maze)
- Immunohistochemistry supplies (anti-Aβ antibodies)
Procedure:
- In Vitro Aggregation Kinetics:
- Prepare Aβ42 peptide solution (50 μM) in PBS.
- Add various concentrations of L-arginine (0.1-10 mM) to the Aβ42 solution.
- Add Thioflavin T (5 μM) to monitor aggregation.
- Measure fluorescence (excitation 440 nm, emission 485 nm) every 10 minutes for 24-48 hours.
- Calculate aggregation half-time and maximum fluorescence for each condition.
In Vivo Efficacy in Mouse Model:
- Administer L-arginine (100-400 mg/kg/day) in drinking water to AppNL-G-F mice starting at 3 months of age for 3-6 months.
- Perform behavioral assessments (Morris water maze for spatial memory, Y-maze for working memory) monthly.
- Euthanize mice and collect brain tissue for analysis.
- Quantify Aβ plaque burden using immunohistochemistry and image analysis software.
- Measure insoluble Aβ42 levels via ELISA.
Neuroinflammation Assessment:
- Isolate RNA from brain tissue.
- Perform qRT-PCR for pro-inflammatory cytokine genes (TNF-α, IL-1β, IL-6).
Expected Outcomes: Effective chemical chaperones should delay Aβ aggregation in vitro, reduce plaque burden and neuroinflammation in mouse brains, and correlate with improved cognitive performance in behavioral tests [94].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Chaperone-Targeted Research
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| HSP90 Inhibitors | 17-AAG, NXD30001, PU-H71 | Oncology research | Inhibit HSP90 ATPase activity; degrade oncogenic client proteins [90] [93] |
| HSP70 Modulators | Pifithrin-μ, VER-155008, JG-98 | Oncology & neurodegeneration research | Inhibit HSP70 function; induce apoptosis or reduce protein aggregation [90] [93] |
| Chemical Chaperones | Arginine, 4-PBA | Neurodegeneration research | Stabilize protein folding; reduce aggregation of amyloidogenic proteins [94] |
| sHSP Inhibitors | NA49 | Oncology research | Disrupt HSP27 oligomerization; sensitive to chemotherapy [93] |
| siRNA Libraries | HSPA5/GRP78, HSP70, HSP90 | Target validation | Genetically validate chaperone targets via knockdown studies [90] |
| Chaperone Antibodies | Anti-HSP90, anti-HSP70, anti-HSP27, anti-HSPA5/GRP78 | Immunodetection | Detect chaperone expression and localization via Western blot, IHC, flow cytometry [90] |
Visualizing Key Pathways and Workflows
Diagram 1: A generalized experimental workflow for evaluating chaperone-targeting therapeutics, progressing from in vitro screening to in vivo validation.
Diagram 2: Disease pathways resulting from chaperone dysregulation, highlighting distinct mechanisms in cancer versus neurodegenerative diseases.
The strategic targeting of molecular chaperones represents a paradigm shift in therapeutic development for complex diseases like cancer and neurodegenerative disorders. The continued elucidation of chaperone structures and mechanisms, coupled with innovative drug design approaches, is yielding increasingly sophisticated therapeutic candidates [33]. Future directions will likely see greater emphasis on isoform-selective inhibitors, targeted disruption of specific protein-protein interactions within the chaperone network, and multi-specific molecules that simultaneously modulate multiple components of the proteostasis network [33] [93]. The promising results from chemical chaperones like arginine in Alzheimer's models highlight the potential for cost-effective, rapidly translatable therapies [94]. As our understanding of the chaperone system deepens, the journey from bench to bedside for chaperone-targeted therapies will undoubtedly accelerate, offering new hope for treating some of the most challenging human diseases.
Conclusion
Chaperone-assisted refolding is a powerful and indispensable methodology that mirrors the sophistication of cellular protein quality control. Mastering the foundational mechanisms, robust protocols, and rigorous validation techniques enables researchers to successfully recover functional proteins from denatured states, a critical capability for biotechnology and therapeutic development. The ongoing structural elucidation of chaperone-client complexes and the development of novel chemical and pharmacological chaperones are paving the way for groundbreaking applications. Future research will undoubtedly focus on engineering chaperones for specific industrial processes and developing targeted therapies that manipulate chaperone function to treat a wide array of protein misfolding diseases, from cancer to neurodegeneration. The integration of these advanced refolding strategies will continue to accelerate discovery in both basic science and clinical research.
References
- 1. Solution of Levinthal's Paradox and a Physical Theory of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7072185/]
- 2. Levinthal's paradox [https://en.wikipedia.org/wiki/Levinthal%27s_paradox]
- 3. Navigating the landscape of protein folding and proteostasis [https://www.nature.com/articles/s41392-025-02439-w]
- 4. How proteins manage to fold and how chaperones ... [https://www.sciencedirect.com/science/article/abs/pii/S1571064524001751]
- 5. Quantitative and scalable approaches for measuring protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9337709/]
- 6. Article Role of Hsp70 chaperone in client-protein folding ... [https://www.sciencedirect.com/science/article/pii/S000634952500534X]
- 7. Energy landscape underlying spontaneous insertion and ... [https://www.nature.com/articles/s41467-018-07320-9]
- 8. A comprehensive application of FiveFold for conformation ... [https://www.nature.com/articles/s41598-025-17022-0]
- 9. Molecular Chaperonin HSP60: Current Understanding and ... [https://pubmed.ncbi.nlm.nih.gov/38791521/]
- 10. Hsp70 chaperones: Cellular functions and molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2773841/]
- 11. The Protective and Therapeutic Function of Small Heat Shock ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3342061/]
- 12. Role of Hsp70 Chaperone in Client-Protein Folding ... [https://pubmed.ncbi.nlm.nih.gov/40873036/]
- 13. Hsf1 and the molecular chaperone Hsp90 support a ' ... [https://pubmed.ncbi.nlm.nih.gov/38059913/]
- 14. Molecular Chaperonin HSP60: Current Understanding and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121636/]
- 15. HSP60 possesses a GTPase activity and mediates protein ... [https://www.nature.com/articles/s41598-017-17167-7]
- 16. Molecular chaperone function of three small heat-shock ... [https://www.sciencedirect.com/science/article/pii/S1355814523000810]
- 17. Small heat shock proteins operate as molecular ... [https://www.nature.com/articles/s41556-022-01074-9]
- 18. The interactions of molecular chaperones with client proteins [https://pmc.ncbi.nlm.nih.gov/articles/PMC8567204/]
- 19. Conformational selection in substrate recognition by Hsp70 chaperones [https://pubmed.ncbi.nlm.nih.gov/23207294/]
- 20. Conserved conformational selection mechanism of Hsp70... [https://elifesciences.org/articles/32764]
- 21. Multifunctional synthetic nano-chaperone for peptide ... [https://www.nature.com/articles/s41467-022-32268-2]
- 22. Refolding Techniques for Recovering Biologically Active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4030991/]
- 23. Quantitative analysis of chaperone network throughput in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3791555/]
- 24. Trigger factor in complex with the ribosome forms a ... [https://www.nature.com/articles/nature02899]
- 25. Resolving chaperone-assisted protein folding on the ... [https://www.nature.com/articles/s41594-024-01355-x]
- 26. Versatility of Trigger Factor Interactions with Ribosome ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2934658/]
- 27. Mechanism of chaperone coordination during ... [https://pubmed.ncbi.nlm.nih.gov/38908370/]
- 28. Arrest Peptide Profiling resolves co-translational folding ... [https://www.nature.com/articles/s41467-025-61398-6]
- 29. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 30. ATP-Independent Chaperones [https://pubmed.ncbi.nlm.nih.gov/35167761/]
- 31. ATP-Driven Molecular Chaperone Machines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3814418/]
- 32. Protein plasticity underlines activation and function of ATP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4516975/]
- 33. Advances in the structures, mechanisms and targeting of ... [https://www.nature.com/articles/s41392-025-02166-2]
- 34. Mechanism of the small ATP-independent chaperone Spy ... [https://www.nature.com/articles/s41467-021-21120-8]
- 35. Strategies for the recovery of active proteins through refolding ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/1475-2859-3-11]
- 36. Optimization of Methods for the Production and Refolding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7551518/]
- 37. Protein Aggregation in vitro and in vivo: A Quantitative ... [https://www.nature.com/articles/nbt0991-825]
- 38. Quantitative analytics for protein refolding states [https://www.sciencedirect.com/science/article/abs/pii/S1359511323003811]
- 39. Quantitative evaluation of refolding conditions for a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3104239/]
- 40. Refolding Proteins Made Easy: 21 Tips and Tricks [https://bitesizebio.com/59217/refolding-proteins-tips-tricks/]
- 41. Catalyzing Protein Folding by Chaperones - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12562196/]
- 42. Artificial Chaperone-assisted Refolding of Carbonic ... [https://www.sciencedirect.com/science/article/pii/S0021925817458844]
- 43. Artificial chaperone-assisted refolding of proteins [https://pubmed.ncbi.nlm.nih.gov/9556524/]
- 44. and beta-cyclodextrin assisted refolding of SDS-induced ... [https://www.sciencedirect.com/science/article/abs/pii/S0167732224021317]
- 45. Optimizing protein folding in prokaryotes: Strategies to ... [https://www.sciencedirect.com/science/article/abs/pii/S0960852425012337]
- 46. Exploiting cyclodextrins as artificial chaperones to enhance ... [https://pubs.rsc.org/en/content/articlelanding/2024/nr/d3nr06044f]
- 47. for Spontaneous and Chaperonin- Protocol in vitro assisted ... Refolding [https://pmc.ncbi.nlm.nih.gov/articles/PMC8329468/]
- 48. Faster and more efficient folding by eliminating a cis–trans ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3970891/]
- 49. Osmolyte-Induced Folding and Stability of Proteins [https://pmc.ncbi.nlm.nih.gov/articles/PMC7393045/]
- 50. Keeping Your Proteins Happy with Chemical Chaperones [https://bitesizebio.com/22039/keeping-your-proteins-happy-with-chemical-chaperones/]
- 51. A proteome-wide map of chaperone - assisted in... protein refolding [https://pubmed.ncbi.nlm.nih.gov/36417429/]
- 52. Role of the chaperonin TCP-1 ring complex in protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12277325/]
- 53. Aggregation Prevention Assay for Chaperone Activity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8376500/]
- 54. A Proteome-Wide Map of Chaperone - Assisted in... Protein Refolding [https://colab.ws/articles/10.1101%2F2021.11.20.469408]
- 55. Exploring Protein Aggregation in Biological Products [https://link.springer.com/article/10.1208/s12249-025-03189-2]
- 56. Direct observation of kinetic traps associated with structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3420203/]
- 57. Protein folding: Defining a “standard” set of experimental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2279278/]
- 58. Chaperone-mediated protein folding [https://pubmed.ncbi.nlm.nih.gov/10221986/]
- 59. Chaperone (protein) [https://en.wikipedia.org/wiki/Chaperone_(protein)]
- 60. Protein folding while chaperone bound is dependent on ... [https://www.nature.com/articles/s41467-019-12774-6]
- 61. Catalyzing Protein Folding by Chaperones [https://www.mdpi.com/2079-7737/14/10/1450]
- 62. Chaperone dependency during biogenesis does not ... [https://link.springer.com/article/10.1038/s44320-025-00166-6]
- 63. Visualizing chaperone-assisted protein folding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4937829/]
- 64. A proteome-wide map of chaperone-assisted protein refolding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9860312/]
- 65. emerging roles of GrpE-like NEFs in proteostasis and ... [https://pubmed.ncbi.nlm.nih.gov/41134158/]
- 66. A quantitative chaperone interaction network reveals the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4104544/]
- 67. Engineering and Evolution of Molecular Chaperones ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2016.00008/full]
- 68. Hsp90 co-chaperone FKBP4 facilitates CCT8 folding and ... [https://www.sciencedirect.com/science/article/pii/S0021925825027668]
- 69. A Systematic Protein Refolding Screen Method using the ... [https://www.nature.com/articles/s41598-017-09687-z]
- 70. Protocol for preparing proteins with improved solubility by ... [https://pubmed.ncbi.nlm.nih.gov/17948006/]
- 71. Refolding of chemically denatured α-amylase in dilution ... [https://www.sciencedirect.com/science/article/abs/pii/S030441650400162X]
- 72. An automated in vitro protein folding screen applied to a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2286713/]
- 73. Artificial chaperone-assisted refolding of carbonic anhydrase B [https://pubmed.ncbi.nlm.nih.gov/8631951/]
- 74. Mechanistic comparison of artificial-chaperone-assisted ... [https://pubmed.ncbi.nlm.nih.gov/9889157/]
- 75. -Based Fluorescence : Ascertaining the... Enzyme Activity Assay [https://pubmed.ncbi.nlm.nih.gov/39062935/]
- 76. Top Enzymatic Assays for Drug Screening in 2025 [https://synapse.patsnap.com/article/top-enzymatic-assays-for-drug-screening-in-2025]
- 77. First complete structures of heat shock chaperone protein ... [https://www.stjude.org/media-resources/news-releases/2025-medicine-science-news/first-complete-structures-of-heat-shock-chaperone-protein-complex-reveal-handoff-mechanism.html]
- 78. From denaturant to ribosome: rethinking chaperone ... [https://link.springer.com/article/10.1038/s44320-025-00167-5]
- 79. Measurement of Fluorescence of CheY Autophosphorylation... Kinetics [https://experiments.springernature.com/articles/10.1007/978-1-4939-7577-8_25?error=cookies_not_supported&code=34b9d7ad-720d-4f41-ab56-a6b8a9d3d56f]
- 80. Hydrogen deuterium exchange mass spectrometry applied to... [https://pubmed.ncbi.nlm.nih.gov/31215268/]
- 81. Quantitative Evaluation of Native Protein Folds and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6318237/]
- 82. Resolving chaperone - assisted folding on the ribosome at the... protein [https://www.nature.com/articles/s41594-024-01355-x?error=cookies_not_supported&code=72f00851-3f8b-42f7-820e-4c1466ac60cb]
- 83. Strategies for the recovery of active proteins through refolding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC517725/]
- 84. Pathway regulation mechanism by cotranslational protein ... [https://www.nature.com/articles/s42004-025-01636-6]
- 85. Kinetic and structural comparison of a protein 's cotranslational ... [https://pubmed.ncbi.nlm.nih.gov/29854950/]
- 86. Regulation of molecular chaperones through post- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4492711/]
- 87. Chaperone code [https://en.wikipedia.org/wiki/Chaperone_code]
- 88. Pathway regulation mechanism by cotranslational protein ... [https://pubmed.ncbi.nlm.nih.gov/40750991/]
- 89. FoldPAthreader: predicting protein folding pathway using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11167914/]
- 90. Beneficial Handling of Molecular Chaperones ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468336/]
- 91. Chaperones as Potential Pharmacological Targets for ... [https://pubmed.ncbi.nlm.nih.gov/39871559]
- 92. Targeting protein misfolding in Alzheimer's disease [https://www.sciencedirect.com/science/article/pii/S0753332225007255]
- 93. Emerging opportunities and challenges in small molecule ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425008657]
- 94. Simple amino acid supplement greatly reduces Alzheimer's ... [https://www.sciencedaily.com/releases/2025/11/251121090731.htm]