ATP-Driven Protein Folding: The Molecular Mechanism and Biomedical Applications of Chaperonins
This article provides a comprehensive, state-of-the-art analysis of the ATP-dependent mechanism of chaperonins, essential molecular machines that facilitate protein folding in the cell.
ATP-Driven Protein Folding: The Molecular Mechanism and Biomedical Applications of Chaperonins
Abstract
This article provides a comprehensive, state-of-the-art analysis of the ATP-dependent mechanism of chaperonins, essential molecular machines that facilitate protein folding in the cell. Aimed at researchers and drug development professionals, it begins by exploring the foundational structural biology and energy transduction principles of Group I (GroEL/GroES) and Group II chaperonins. It then details cutting-edge methodologies for studying their ATPase cycles and applications in biotechnology. The article further addresses common experimental challenges and optimization strategies for in vitro assays. Finally, it validates these mechanisms through comparative analysis with other chaperone systems and discusses emerging therapeutic targeting in protein misfolding diseases. This synthesis of mechanistic insight and practical application serves as a critical resource for advancing fundamental knowledge and developing novel biomedical interventions.
Unfolding the Machine: Core Principles and Structures of ATP-Powered Chaperonins
This technical guide, framed within a broader thesis on ATP-dependent chaperonin mechanisms, provides a definitive classification of chaperonin systems. Chaperonins are essential molecular machines that facilitate protein folding in an ATP-dependent manner. They are subdivided into two evolutionarily and structurally distinct groups: Group I (exemplified by the bacterial GroEL/GroES system) and Group II (exemplified by the eukaryotic cytosolic chaperonin TRiC/CCT). This document delineates their structural, functional, and mechanistic differences, supported by current quantitative data and experimental methodologies relevant to ongoing research in biomedicine and drug development.
Chaperonins are a class of molecular chaperones that form large, double-ring complexes with a central cavity that provides an isolated environment for protein folding. Their ATP-driven cycle is a cornerstone of cellular proteostasis. The classification into Group I and Group II is based on several key characteristics:
- Group I: Found in eubacteria (GroEL/GroES), eukaryotic organelles of prokaryotic origin (mitochondria, chloroplasts), and some archaea. They require a separate, detachable "lid" co-chaperonin (e.g., GroES).
- Group II: Found in the eukaryotic cytosol (TRiC/CCT) and in archaea (thermosome). They feature a built-in lid structure and have hetero-oligomeric subunits with specific substrate-binding properties.
Structural and Functional Comparison
The core distinctions between the two groups are summarized in the table below.
Table 1: Comparative Analysis of Group I and Group II Chaperonins
| Feature | Group I (GroEL/GroES) | Group II (TRiC/CCT) |
|---|---|---|
| Organismic Distribution | Eubacteria, Eukaryotic Organelles (Mitochondria, Chloroplasts) | Eukaryotic Cytosol, Archaea |
| Prototypical System | GroEL (chaperonin) + GroES (co-chaperonin) | TRiC (TCP-1 Ring Complex) / CCT (Chaperonin Containing TCP-1) |
| Ring Composition | Homo-oligomeric (7 identical subunits per ring) | Hetero-oligomeric (8 distinct but homologous subunits per ring: CCT1-8) |
| Lid Mechanism | Detachable co-chaperonin (GroES) forms a separate mobile lid. | Built-in, helical protrusions form an integrated, non-detachable lid. |
| ATPase Activity | Positive intra-ring cooperativity; negative inter-ring cooperativity. | More complex, subunit-specific ATPase activities with sequential firing. |
| Substrate Specificity | Broad, hydrophobic-rich peptides and misfolded proteins. | High specificity for structurally complex proteins (e.g., actins, tubulins, cell cycle regulators). |
| Folding Chamber | Hydrophilic chamber wall, encapsulation provides an "Anfinsen cage." | Chemically diverse, subunit-specific chamber wall aids in coordinated folding. |
| Key References | (Horwich et al., Cell, 2007; Xu et al., Nature, 1997) | (Yebenes et al., Nat. Struct. Mol. Biol., 2011; Leitner et al., Cell, 2012) |
ATP-Dependent Mechanism: A Comparative Workflow
The ATP-driven cycles of both groups share a common principle—coordinated conformational changes triggered by ATP binding and hydrolysis—but differ in execution.
Experimental Protocol 1: Monitoring the ATPase Cycle Using Coupled Enzymatic Assay
- Objective: Quantify ATP hydrolysis rates of chaperonin complexes.
- Methodology: A continuous spectrophotometric assay is used. The hydrolysis of ATP to ADP and inorganic phosphate (Pi) is coupled to the oxidation of NADH, which is monitored by a decrease in absorbance at 340 nm.
- Key Steps:
- Purify chaperonin complex (e.g., GroEL, TRiC) to homogeneity via affinity and size-exclusion chromatography.
- Prepare reaction mix containing assay buffer (e.g., 50 mM HEPES-KOH, pH 7.4, 100 mM KCl, 10 mM MgAcetate), 2 mM phosphoenolpyruvate, 0.2 mM NADH, 5 U/mL pyruvate kinase, 5 U/mL lactate dehydrogenase.
- Initiate reaction by adding ATP (typically 1-5 mM final concentration) to the mix containing chaperonin (0.1-1 µM complex).
- Record absorbance at 340 nm for 10-30 minutes using a spectrophotometer with a temperature-controlled cuvette holder (25-37°C).
- Calculate ATPase rate using the extinction coefficient for NADH (ε340 = 6220 M⁻¹cm⁻¹).
Diagram 1: Generalized ATP-Driven Chaperonin Cycle (Max Width: 760px)
Diagram 2: Side-by-Side Comparison of Group I & II Mechanisms (Max Width: 760px)
Experimental Protocols for Functional Study
Experimental Protocol 2: Substrate Folding Assay Using Native Gel Electrophoresis
- Objective: Assess the ability of chaperonins to fold denatured substrates into their native conformation.
- Methodology: Monitor the recovery of native structure of a model substrate (e.g., mitochondrial malate dehydrogenase, MDH for GroEL; actin for TRiC) after chaperonin-assisted refolding.
- Key Steps:
- Substrate Denaturation: Denature purified substrate protein (50 µM) in 6 M Guanidine-HCl, 50 mM Tris-HCl (pH 7.5), 20 mM DTT for 1 hour at 25°C.
- Refolding Initiation: Rapidly dilute the denatured substrate 100-fold into refolding buffer (50 mM HEPES-KOH, pH 7.4, 50 mM KCl, 10 mM MgCl2, 2 mM DTT) containing:
- Experimental: Chaperonin complex (1 µM) and ATP (2 mM).
- Controls: Buffer only (spontaneous refolding), Chaperonin + non-hydrolyzable ATP analog (e.g., ATPγS).
- Incubation: Allow refolding to proceed at 25°C for 60-90 minutes.
- Analysis: Stop the reaction with native gel loading buffer (no SDS). Resolve samples on a non-denaturing polyacrylamide gel (e.g., 6-10%). Native protein migrates distinctly from unfolded/aggregated material, visualized by Coomassie staining or immunoblotting.
Experimental Protocol 3: Cryo-EM Workflow for Structural Analysis of Chaperonin States
- Objective: Obtain high-resolution structures of chaperonin complexes in different nucleotide states.
- Methodology: Single-particle cryo-electron microscopy.
- Key Steps:
- Sample Preparation: Incubate purified chaperonin (3-5 mg/mL) with desired nucleotide (e.g., 5 mM ATP, ADP, or AMP-PNP) and optionally, substrate protein or co-chaperonin. Apply 3-4 µL to a glow-discharged cryo-EM grid, blot, and plunge-freeze in liquid ethane.
- Data Collection: Acquire movie micrographs on a 300 keV cryo-TEM with a direct electron detector (e.g., Gatan K3, Falcon 4). Target a defocus range of -1.0 to -2.5 µm. Collect 3000-5000 micrographs.
- Image Processing: Motion correction and dose-weighting (e.g., MotionCor2). Particle picking (e.g., crYOLO, Relion). 2D classification to remove junk particles. Multiple rounds of 3D classification in Relion or CryoSPARC to separate conformational states. Non-uniform refinement to obtain final high-resolution maps.
- Model Building: Fit existing high-resolution structures (e.g., PDB IDs) into the map using rigid-body fitting in Chimera or Coot, followed by iterative real-space refinement in Phenix.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Chaperonin Research
| Item | Function in Research | Example Product/Catalog # (for illustration) |
|---|---|---|
| Purified Chaperonin Complexes | Essential substrate for all biochemical/structural studies. | Recombinant GroEL/GroES (Sigma-Aldrich, CH0405); Bovine TRiC/CCT ( homemade via affinity tags). |
| Non-Hydrolyzable ATP Analogs | To trap and study specific nucleotide-bound states of the cycle. | Adenosine 5'-(γ-thio)-triphosphate (ATPγS, Roche, 11162306001); AMP-PNP (Sigma, A2647). |
| Model Folding Substrates | Well-characterized proteins to assay chaperonin function. | For GroEL: Rhodanese (Roche, 108198), Malate Dehydrogenase (Sigma, M2634). For TRiC: Firefly Luciferase (Promega, E1701), β-Actin (Cytoskeleton, AKL99). |
| Coupled ATPase Assay Kit | Convenient system for continuous monitoring of ATP hydrolysis kinetics. | ATPase/GTPase Activity Assay Kit (Sigma, MAK113). |
| Crosslinking Reagents | To capture transient interactions between chaperonin, lid, and substrate. | BS³ (Thermo Fisher, 21580); Glutaraldehyde (Electron Microscopy Sciences, 16220). |
| Cryo-EM Grids | Supports for vitrified sample in single-particle analysis. | Quantifoil R1.2/1.3 Au 300 mesh grids (Electron Microscopy Sciences, Q350AR13A). |
| Negative Stain Reagents | For rapid, initial assessment of chaperonin sample quality and homogeneity. | Uranyl Acetate (2% solution, Electron Microscopy Sciences, 22400). |
| Size-Exclusion Columns | Final polishing step to obtain monodisperse, aggregation-free chaperonin complexes. | Superose 6 Increase 10/300 GL (Cytiva, 29091596). |
The elucidation of ATP-dependent mechanisms in chaperonin research is fundamentally reliant on a precise understanding of the GroEL/GroES complex's architecture. This double-ring complex, a member of Group I chaperonins, facilitates protein folding through concerted, ATP-driven conformational changes. This whitepaper provides an in-depth structural and functional analysis of its domains within the context of modern mechanistic research.
Structural Domains and Quantitative Parameters
The GroEL double-ring complex comprises 14 identical subunits arranged in two heptameric rings stacked back-to-back. Each subunit is organized into discrete functional domains.
Table 1: Quantitative Structural Parameters of the GroEL/GroES Complex
| Parameter | Value (Wild-type E. coli GroEL) | Description/Implication |
|---|---|---|
| Oligomeric State | 14 subunits (2 rings of 7) | Forms the core double-ring architecture. |
| Molecular Mass | ~800 kDa (GroEL alone) | Large size necessitates cryo-EM for structural analysis. |
| Ring Diameter | ~137 Å | Defines the central cavity dimensions. |
| Cavity Diameter (Apo) | ~45 Å | Restrictive size in the trans ring. |
| Cavity Diameter (Bulged) | ~60-65 Å | Expanded size in the GroES-capped cis ring. |
| ATPase Sites per Ring | 7 (one per subunit) | Cooperativity within a ring is positive; between rings is negative. |
| ATP Hydrolysis Rate (per ring) | ~12 s⁻¹ (at 25°C) | Kinetics are central to the folding cycle timing. |
| K~D~ for GroES | ~1 µM (in presence of ATP) | High-affinity binding during the functional cycle. |
Table 2: Core Structural Domains of a GroEL Subunit
| Domain | Residue Range (approx.) | Primary Function | Key Structural Motifs |
|---|---|---|---|
| Equatorial | 1-137, 409-523 | ATP binding/hydrolysis; inter-subunit & inter-ring contacts. | Nucleotide-binding pocket, ring-ring interface helices. |
| Intermediate | 138-190, 377-408 | Hinge region; transmits ATP signal to apical domain. | Flexible hinges, conserved glycine residues. |
| Apical | 191-376 | Substrate (unfolded protein) & GroES co-chaperonin binding. | Hydrophobic lining (HPD loop, helix H, I), mobile helices. |
Experimental Protocols for Domain Analysis
Cryo-Electron Microscopy (cryo-EM) for Structural State Determination
This protocol resolves different conformational states (apo, ATP-bound, GroES-capped).
Protocol:
- Sample Preparation: Purify GroEL (and GroES/substrate if needed) to homogeneity. Incubate with 1mM ATPγS (non-hydrolyzable analog) and a 2-fold molar excess of GroES to trap the cis-folding complex.
- Vitrification: Apply 3.5 µL of sample (~3 mg/mL) to a glow-discharged holey carbon grid. Blot for 3-5 seconds under 100% humidity at 4°C and plunge-freeze in liquid ethane using a vitrification device.
- Data Acquisition: Collect images on a 300 keV cryo-TEM with a K3 direct electron detector. Use a nominal magnification of 105,000x (pixel size 0.826 Å). Collect a dose-framed movie series with a total dose of 50 e⁻/Ų over 40 frames.
- Image Processing: Motion-correct and dose-weight frames. Pick particles automatically. Perform 2D classification to remove junk particles. Use an initial 3D model for heterogeneous refinement in Relion or cryoSPARC to separate structural states (e.g., apo, bullet-shaped, football-shaped complexes).
- Model Building & Refinement: Fit existing high-resolution X-ray structures of domains into the cryo-EM density map using Chimera. Refine domains and side chains iteratively in Coot and Phenix.
Site-Directed Mutagenesis & ATPase Activity Assay
This protocol probes the functional role of specific domain residues.
Protocol:
- Mutagenesis: Design primers to introduce point mutations (e.g., D398A in equatorial domain to disrupt ATP hydrolysis). Perform PCR using plasmid containing the groEL gene as template. Digest parental DNA with DpnI, transform into competent E. coli, and sequence-verify clones.
- Protein Expression & Purification: Express mutant GroEL in an E. coli strain lacking endogenous chaperonins. Purify via anion-exchange and size-exclusion chromatography.
- ATPase Assay: Use a coupled enzyme system (pyruvate kinase/lactate dehydrogenase) monitoring NADH oxidation at 340 nm. In a 96-well plate, mix 0.5 µM GroEL (wild-type or mutant) in assay buffer (50 mM HEPES-KOH pH 7.5, 10 mM MgCl₂, 25 mM KCl) with 2 mM phosphoenolpyruvate, 0.2 mM NADH, and 5 units/mL of coupling enzymes. Initiate reaction with 2 mM ATP. Record absorbance at 340 nm every 20s for 30 minutes. Calculate hydrolysis rate from the linear decrease.
Visualizing the Chaperonin Functional Cycle
Diagram Title: The ATP-Driven GroEL/GroES Functional Cycle
Diagram Title: GroEL Subunit Domain Architecture and Functions
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Chaperonin Domain & Mechanism Studies
| Reagent/Category | Example(s) | Primary Function in Research |
|---|---|---|
| Chaperonin Proteins | Wild-type GroEL, GroES (from E. coli); Thermosome (from archaea). | Core subjects for structural, biochemical, and functional studies. |
| Site-Directed Mutants | D398A (ATPase-deficient), R231A (substrate binding-deficient), AAG (all-alanine apical). | To dissect the role of specific domains/residues in the ATPase cycle. |
| Nucleotide Analogs | ATPγS, AMP-PNP (non-hydrolyzable); ADP·AlFx (transition state mimic). | To trap and study specific conformational states for structural analysis. |
| Model Substrates | Rhodanese, MDH (Mitochondrial Malate Dehydrogenase), α-Lactalbumin. | Well-characterized, aggregation-prone proteins to assay folding activity. |
| Coupling Enzyme Assay Kits | Pyruvate Kinase/Lactate Dehydrogenase coupled system. | To continuously monitor ATP hydrolysis kinetics by purified chaperonins. |
| Chemical Chaperones/Inhibitors | BeF~3~- (stabilizes ADP state), MDP (Methylene Diphosphonate). | Probe specific steps in the nucleotide cycle and inhibit function. |
| Fluorescent Probes | Bis-ANS, 1,1'-Bi(4-anilino)naphthalene-5,5'-disulfonic acid. | Binds hydrophobic surfaces; reports on substrate binding/release and cavity exposure. |
| Crosslinkers | BS³ (Amine-reactive), EDC (Carbodiimide, for zero-length). | To capture transient intra- or inter-ring interactions for structural mapping. |
| Cryo-EM Grids & Supplies | Quantifoil R1.2/1.3 Au 300 mesh grids; liquid ethane/propane. | For high-resolution sample vitrification and structural state determination. |
This whitepaper details the central role of ATP hydrolysis in powering the chaperonin folding cycle, a core mechanism within a broader thesis on ATP-dependent chaperonin research. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are essential molecular machines that facilitate the correct folding of nascent or misfolded polypeptides. Their function is strictly contingent upon the binding and hydrolysis of adenosine triphosphate (ATP), which provides the conformational energy required for the cyclic entrapment, folding, and release of substrate proteins.
The Thermodynamic Imperative and Hydrolytic Mechanism
Protein folding in the crowded cellular environment is prone to aggregation. Chaperonins overcome this by providing an isolated Anfinsen cage, the conformational changes of which are driven by ATP. The hydrolysis of ATP to ADP and inorganic phosphate (Pi) releases approximately -30.5 kJ/mol under standard conditions, but this standard free energy of hydrolysis is significantly magnified in-cell due to maintained high ATP:ADP ratios.
Table 1: Quantitative Parameters of ATP Hydrolysis in Model Chaperonins
| Parameter | GroEL/GroES (E. coli) | TRiC/CCT (Human) | Notes / Experimental Method |
|---|---|---|---|
| ATP Molecules per Cycle | 7 per ring; 14 total | 8 per ring; 16 total | Measured via stoichiometric binding assays and cryo-EM. |
| Intrinsic Hydrolysis Rate (k~hyd~) | ~1.2 min⁻¹ per site | ~0.8 min⁻¹ per site | Determined by quench-flow experiments using [γ-³²P]ATP. |
| Cooperative Hill Coefficient | ~2-4 (positive within ring) | ~1.5-3 (positive within ring) | Derived from ATPase activity vs. [ATP] plots. Negative cooperativity between rings. |
| ΔG~hydrolysis~ (in-cell estimate) | ~ -50 to -60 kJ/mol | ~ -50 to -60 kJ/mol | Calculated using measured cellular [ATP], [ADP], [Pi]. |
| Major Conformational Change Trigger | ATP binding (T→R state) | ATP binding & hydrolysis | Monitored by time-resolved FRET and cryo-EM. |
| Folding Cycle Time | ~10-15 seconds | ~20-30 seconds | Measured via single-molecule FRET and stopped-flow techniques. |
The hydrolytic mechanism involves nucleophilic attack by a water molecule on the γ-phosphate of ATP, facilitated by Mg²⁺ coordination and key catalytic residues (e.g., Asp⁸⁷ in GroEL). The release of Pi is often the rate-limiting step and is tightly coupled to the massive quaternary structural shifts of the chaperonin complex.
Detailed Experimental Protocols
Protocol: Measuring ATPase Kinetics of Chaperonins Using a Coupled Enzymatic Assay
Objective: To determine the steady-state kinetics (K~M~, V~max~) of chaperonin ATP hydrolysis.
Materials:
- Purified chaperonin complex (e.g., GroEL, TRiC).
- ATP, phospho(enol)pyruvate (PEP), NADH.
- Pyruvate kinase (PK) and lactate dehydrogenase (LDH).
- Assay buffer (typically 50 mM HEPES-KOH pH 7.4, 100 mM KCl, 10 mM MgCl₂).
- Spectrophotometer or plate reader capable of monitoring absorbance at 340 nm.
Procedure:
- Prepare a master mix containing assay buffer, 2 mM PEP, 0.2 mM NADH, 20 U/ml PK, and 20 U/ml LDH.
- In a cuvette or plate well, add master mix and chaperonin to a final concentration of 50-100 nM (as a hexadecamer).
- Initiate the reaction by adding ATP across a concentration range (e.g., 0.01 to 2 mM).
- Immediately monitor the decrease in absorbance at 340 nm (A₃₄₀) for 5-10 minutes. The oxidation of NADH to NAD⁺ is stoichiometric with ATP hydrolyzed (ATP → ADP + Pi; PEP + ADP → Pyruvate + ATP; Pyruvate + NADH + H⁺ → Lactate + NAD⁺).
- Calculate the rate of ATP hydrolysis from the linear slope of A₃₄₀ vs. time, using the extinction coefficient for NADH (ε₃₄₀ = 6220 M⁻¹cm⁻¹).
- Plot hydrolysis rate vs. [ATP] and fit data to the Michaelis-Menten equation to derive K~M~ and V~max~.
Protocol: Single-Round ATP Hydrolysis Quench-Flow Experiment
Objective: To measure the intrinsic rate of ATP hydrolysis (k~hyd~) and Pi release, disentangling binding from hydrolysis.
Materials:
- Purified chaperonin.
- [γ-³²P]ATP (high specific activity).
- Quench-flow apparatus.
- Quench solution (5 M formic acid, 0.5 M EDTA).
- Charcoal slurry (5% w/v in 50 mM NaH₂PO₄).
Procedure:
- Pre-incubate chaperonin (1 µM complex) in assay buffer at 25°C.
- Rapidly mix with an equal volume of [γ-³²P]ATP (2 µM final, ~500 cpm/pmol) in the quench-flow apparatus.
- Allow the reaction to proceed for varying time intervals (ms to s) before quenching with formic acid/EDTA.
- The acid quench denatures the protein and stops the reaction. Centrifuge the sample.
- Separate hydrolyzed Pi from unhydrolyzed ATP by adding supernatant to activated charcoal slurry, which binds nucleotide. Centrifuge.
- Quantify ³²P~i~ in the supernatant (released Pi) via liquid scintillation counting.
- Plot Pi released vs. time and fit to a single or double exponential to obtain the rate constant(s) for hydrolysis.
Visualizing the Chaperonin ATP-Driven Cycle
Diagram 1: GroEL/ES ATP-Driven Folding Cycle (78 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Studying ATP-Dependent Chaperonin Mechanisms
| Reagent / Material | Primary Function in Research | Example & Notes |
|---|---|---|
| Purified Chaperonin Complex | The core macromolecule for functional and structural studies. | GroEL₁₄/GroES₇ from E. coli; Recombinant human TRiC/CCT expressed in insect cells. Tagged variants (His, FLAG) for purification. |
| ATP Analogs & Radioisotopes | Probing binding, hydrolysis, and conformational steps. | ATPγS (non-hydrolyzable): Traps ATP-bound states for cryo-EM. [γ-³²P]ATP: Measures hydrolysis kinetics (quench-flow). [α-³²P]ATP: Studies nucleotide exchange. |
| Coupled Enzyme ATPase Assay Kit | Continuous, spectrophotometric measurement of steady-state ATP hydrolysis. | Commercial kits (e.g., Sigma-Aldrich MAK113) provide PK/LDH enzymes, PEP, and NADH for robust, high-throughput activity screens. |
| Fast-Kinetics Apparatus | Resolving pre-steady-state kinetics of binding, hydrolysis, and conformational changes. | Stopped-Flow Spectrometer: Monitors fluorescence/absorbance changes on ms-s timescale. Quench-Flow Instrument: For chemical trapping of intermediates (e.g., Pi release). |
| Fluorescent Nucleotides | Reporting conformational changes via FRET or direct fluorescence. | Mant-ATP (2´(3´)-O-(N-Methylanthraniloyl)adenosine-5´-triphosphate): Environment-sensitive fluorophore used in FRET with protein tryptophans to monitor binding. |
| Conformation-Specific Antibodies | Detecting specific functional states of chaperonins in vitro or in cellulo. | Antibodies raised against GroEL apical domain epitopes exposed only in the ATP-bound (R) state. Useful for Western blot or immunoprecipitation of specific conformers. |
| Refolding Reporter Substrates | Model clients to assay chaperonin folding activity. | Mitochondrial Malate Dehydrogenase (mtMDH): Classic, aggregation-prone substrate. Folding is measured by recovery of enzymatic activity. Fluorescently labeled Rhodanese: Folding monitored by fluorescence recovery upon dilution from denaturant. |
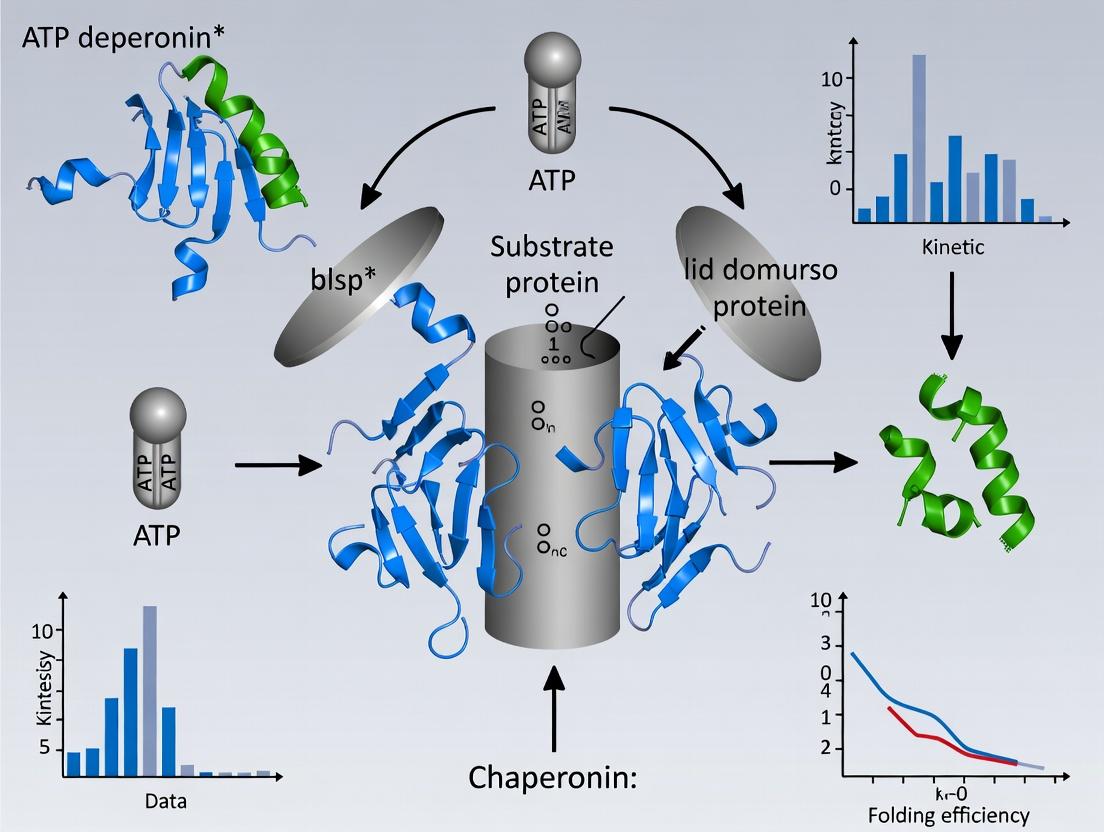
Within the broader thesis on ATP-dependent mechanisms of chaperonin research, the GroEL/GroES system in E. coli and its eukaryotic homolog TRiC/CCT represent paradigmatic models for understanding regulated protein folding. This whitepaper provides an in-depth technical examination of the concerted allosteric cycle governing substrate polypeptide binding, encapsulation within a folding chamber, and subsequent release. The cycle is driven by ATP binding and hydrolysis, coupled with co-chaperonin binding, which orchestrates large conformational changes essential for function.
The Concerted Allosteric Cycle: A Stage-by-Stage Analysis
Stage 1: Initial Substrate Binding to theTransRing
The unfolded or misfolded substrate protein, with exposed hydrophobic residues, binds to the apical domains of the GroEL ring opposite the GroES-bound (cis) ring. Binding is mediated by hydrophobic interactions and is influenced by the nucleotide state (typically ADP-bound in the cis ring, empty in the trans ring).
Stage 2: ATP Binding and Allosteric Priming
ATP binding to the trans ring initiates a concerted conformational change. The seven subunits of the trans ring bind ATP cooperatively, leading to rigid-body elevation and twist of the apical domains. This weakens substrate binding and prepares the ring for GroES engagement.
Stage 3: GroES Encapsulation and Chamber Transformation
GroES binds to the ATP-primed trans ring in a nucleotide-dependent manner, discharging the substrate polypeptide into the now-enlarged cis cavity. The chamber volume increases approximately twofold. The hydrophilic lining of the chamber is revealed, promoting productive folding in isolation.
Stage 4: Folding Clock and ATP Hydrolysis
Productive folding proceeds for a period tied to the ATP hydrolysis rate in the cis ring (~10-15 seconds). Hydrolysis to ADP+Pi relaxes the allosteric constraints, priming the system for the next triggering event.
Stage 5: Asymmetric Trigger of Release
ATP binding to the opposing (trans) ring, now with substrate, serves as the signal for discharge. This binding induces a conformational strain that destabilizes the cis complex (GroES and ADP).
Stage 6: GroES Dissociation and Substrate Ejection
The cis GroES and ADP are released. The substrate, which may be folded, partially folded, or misfolded, is ejected. If not native, it can rebind for another round of encapsulation.
Table 1: Key Quantitative Parameters of the GroEL/ES Concerted Cycle
| Parameter | Value | Experimental Method | Significance |
|---|---|---|---|
| ATP Molecules per Cycle | 7 ATP/cis ring, 14 ATP/full cycle | Isothermal Titration Calorimetry (ITC) | Drives conformational changes & timing |
| Folding Chamber Volume (cis) | ~175,000 ų | Cryo-EM Reconstruction | Determines substrate size limit |
| Folding Time (Encapsulation) | 10-15 seconds | Single-turnover Kinetics | Set by ATP hydrolysis rate |
| GroEL:GroES:ATP Stoichiometry | 1:1:7 (per functional cis complex) | Analytical Ultracentrifugation | Defines functional unit |
| Substrate Size Range | Up to ~60 kDa | Size-Exclusion Studies | Practical operational limit |
Experimental Protocols for Key Analyses
Protocol 1: Measuring ATPase Activity Kinetics (Coupled Enzyme Assay)
Purpose: To determine the rate of ATP hydrolysis by chaperonin, a key parameter regulating the folding cycle.
- Prepare Reaction Mix: In a 1 mL cuvette, combine 50 mM HEPES-KOH (pH 7.6), 10 mM MgCl₂, 2 mM phosphoenolpyruvate, 0.2 mM NADH, 50 μg/mL pyruvate kinase, 50 μg/mL lactate dehydrogenase.
- Initiate Reaction: Add GroEL (0.5-1 μM heptamer) and start reaction by adding ATP (2 mM final concentration).
- Monitor Continuously: Measure absorbance at 340 nm for 5-10 minutes at 25°C. The oxidation of NADH to NAD⁺ causes a decrease in A₃₄₀.
- Calculate Rate: Using the extinction coefficient for NADH (ε₃₄₀ = 6220 M⁻¹cm⁻¹), calculate the rate of ATP hydrolysis. Normalize to mol ATP hydrolyzed/mol GroEL/sec.
Protocol 2: Substrate Encapsulation Assay (Protected from Proteolysis)
Purpose: To demonstrate substrate sequestration within the cis cavity.
- Form Cis Ternary Complex: Incubate GroEL (2 μM), unfolded substrate (e.g., rhodanese, 4 μM), and GroES (4 μM) in buffer with 2 mM ATP for 2 minutes.
- Protease Challenge: Add proteinase K (0.1 mg/mL final) to the mixture. Incubate on ice for 10 minutes.
- Terminate Digestion: Add phenylmethylsulfonyl fluoride (PMSF) to 5 mM.
- Analyze: Run samples on SDS-PAGE alongside controls (substrate without GroEL/ES, substrate with GroEL but no ATP). Protected substrate indicates successful encapsulation.
Protocol 3: Single-Ring Mutant Analysis
Purpose: To dissect the role of inter-ring negative allostery using a GroEL SR1 mutant (single-ring variant that binds GroES but cannot discharge it).
- Purify GroEL SR1: Express and purify the D398A K4E double mutant GroEL heptamer.
- Form Stable Complex: Mix GroEL SR1 (1 μM), unfolded substrate (2 μM), GroES (2 μM), and ATP (2 mM) in folding buffer. Incubate 30 min.
- Assay Folding: For an enzyme substrate (e.g., malate dehydrogenase), take aliquots and assay activity after dilution to prevent rebinding. Compare to wild-type GroEL.
- Interpretation: Trapping in SR1 demonstrates encapsulation is sufficient for folding of some proteins; comparison to WT shows necessity of release mechanism.
Visualizing the Concerted Cycle and Pathways
Title: The Concerted Allosteric Cycle of GroEL/ES
Title: Encapsulation Protection Assay Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Chaperonin Cycle Research
| Reagent / Material | Function & Rationale |
|---|---|
| GroEL/GroES (Wild-type & Mutants e.g., SR1) | Core chaperonin system components. Mutants allow dissection of specific cycle stages. |
| ATPγS (Adenosine 5′-[γ-thio]triphosphate) | Non-hydrolyzable ATP analog used to trap pre-hydrolysis conformational states for structural analysis. |
| Unfolding Substrates (e.g., Rhodanese, MDH) | Model substrates with well-defined refolding assays to quantify chaperonin activity. |
| Pyruvate Kinase / Lactate Dehydrogenase (PK/LDH) Enzymes | Components of the coupled assay system for continuous, real-time measurement of ATPase activity. |
| Proteinase K | Broad-specificity protease used in encapsulation assays to digest non-encapsulated substrate. |
| ANS (1-Anilinonaphthalene-8-sulfonate) | Hydrophobic fluorescent dye used to monitor exposure/burial of hydrophobic surfaces on chaperonin or substrate. |
| Size-Exclusion Chromatography (SEC) Columns (e.g., Superose 6) | To separate and analyze high-molecular-weight chaperonin complexes (e.g., GroEL:GroES:Substrate). |
| Anti-Hydrophobic Patch Antibodies | Specific antibodies targeting apical domain hydrophobic residues to inhibit substrate binding for functional studies. |
This whitepaper details the allosteric mechanisms governing ATP-dependent chaperonin function, a cornerstone of cellular proteostasis. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are large, double-ring ATPases that facilitate protein folding. Their function is driven by highly cooperative ATP binding and hydrolysis within each ring, coupled with negative inter-ring communication. Understanding this allostery is critical for research into protein misfolding diseases and for developing novel therapeutic strategies that target chaperonin networks.
Allosteric Models and Quantitative Principles
The ATP-driven cycle of chaperonins like GroEL is described by the "Nested Model" of allostery, which combines Monod-Wyman-Changeux (MWC) and Koshland-Némethy-Filmer (KNF) principles. Positive intra-ring cooperativity is treated with an MWC model, while negative inter-ring communication follows a KNF-type mechanism.
Table 1: Key Allosteric Parameters for GroEL (from Isothermal Titration Calorimetry & Kinetic Studies)
| Parameter | Value | Description |
|---|---|---|
| Intra-ring Cooperativity (Hill Coefficient, nH) | 2.5 - 3.5 | High positive cooperativity for ATP binding within a single ring. |
| Kd (T-state ring) | ~ 1-5 µM | ATP affinity for a ring in the tense (T), ATP-free state. |
| Kd (R-state ring) | ~ 0.1-0.5 µM | ATP affinity for a ring in the relaxed (R), ATP-bound state. |
| Inter-ring Allosteric Constant (KRR) | ~ 10-2 - 10-3 | Equilibrium constant favoring asymmetric states; indicates strong negative cooperativity between rings. |
| ATP Hydrolysis Rate (kcat) | ~ 15-20 min-1 per ring | Rate constant for ATP hydrolysis once the ring is fully occupied. |
Experimental Protocols for Probing Allostery
Isothermal Titration Calorimetry (ITC) for ATP Binding
Objective: Measure the thermodynamics (Kd, ΔH, ΔS, stoichiometry (n)) and cooperativity of ATP binding to chaperonin rings.
- Sample Preparation: Dialyze GroEL (10-20 µM, per ring) extensively into ITC buffer (e.g., 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 50 mM KCl). Prepare ATP solution in the identical dialysis buffer.
- Instrument Setup: Degas all solutions. Load the cell with GroEL solution. Load the syringe with ATP solution (typically 10-20x the molar concentration of the cell sample).
- Titration: Perform a series of injections (e.g., 2-10 µL each) of ATP into the protein cell at constant temperature (e.g., 25°C). Measure the heat released or absorbed after each injection.
- Data Analysis: Integrate peak areas. Fit the binding isotherm to a cooperative binding model (e.g., sequential or concerted) to extract Kd, nH, ΔH, and n (sites per ring).
Stopped-Flow Fluorescence for Kinetic Allostery
Objective: Resolve the kinetics of intra-ring ATP binding and inter-ring communication.
- Labeling: Introduce a fluorescent probe, e.g., by mutating a residue (like SPC GroEL F44W) to report on conformational changes.
- Rapid Mixing: Load one syringe with labeled GroEL (1 µM, per ring). Load the second syringe with ATP (at varying concentrations, 5-500 µM) in the same buffer.
- Measurement: Rapidly mix equal volumes. Monitor fluorescence change (e.g., tryptophan emission at >320 nm with excitation at 295 nm) on a millisecond timescale.
- Analysis: Fit the time courses to multi-exponential functions. Plot observed rates vs. [ATP] to elucidate the mechanism and identify kinetic intermediates reflecting cooperative binding.
Single-Ring Mutant (SR1) Analysis
Objective: Decouple intra-ring from inter-ring allostery by studying a single-ring variant.
- Construct: Use the GroEL mutant SR1, where the inter-ring contacts are genetically disrupted.
- Comparative Assays: Perform identical ITC and stopped-flow experiments as above on SR1.
- Interpretation: Compare binding isotherms and kinetics of SR1 to wild-type GroEL. The absence of negative inter-ring cooperativity in SR1 manifests as a simpler, hyperbolic ATP binding curve, isolating intra-ring positive cooperativity.
Key Signaling Pathways and Workflows
ATP-Driven Allosteric Cycle of GroEL
Experimental Workflow for Allostery Mapping
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Chaperonin Allostery Research
| Reagent | Function & Rationale |
|---|---|
| GroEL (Wild-Type) | The canonical model chaperonin for allostery studies. Often expressed in E. coli and purified via anion-exchange and size-exclusion chromatography. |
| GroEL Single-Ring Mutant (SR1) | A double-mutant (AAG → DAD at inter-ring interface) that forms stable single rings. Critical for isolating intra-ring allostery. |
| Fluorescent GroEL Variants (e.g., F44W) | Site-specific tryptophan substitution acts as an intrinsic reporter for ATP-induced conformational changes in stopped-flow experiments. |
| Non-Hydrolyzable ATP Analog (e.g., ATPγS, AMP-PNP) | Used in structural studies (X-ray crystallography, Cryo-EM) and binding assays to trap specific allosteric states without cycle progression. |
| High-Purity ATP (Na+ or Mg2+ Salt) | Essential substrate. Must be of the highest purity and pH-adjusted to prevent artifacts in sensitive assays like ITC. |
| Rapid Kinetics Stopped-Flow System | Instrumentation for measuring conformational changes upon ATP binding on millisecond timescales. |
| Isothermal Titration Calorimeter (ITC) | Gold-standard instrument for directly measuring binding affinity, stoichiometry, and thermodynamics in solution. |
| Size-Exclusion Chromatography (SEC) Columns | For final polishing of chaperonin preparations to ensure oligomeric homogeneity (strictly 14-mer for WT). |
Evolutionary Conservation and Variations Across Kingdoms of Life
Thesis Context: This whitepaper is framed within a broader thesis investigating the mechanistic principles and evolutionary divergence of ATP-dependent chaperonins, focusing on their role in protein folding and implications for drug targeting.
ATP-dependent chaperonins are essential molecular machines, facilitating proper protein folding under physiological and stress conditions. Their core structure and ATPase mechanism are evolutionarily conserved, yet significant variations exist across the kingdoms of life—Archaea, Bacteria, and Eukarya. Understanding these conservation patterns and divergences is critical for elucidating fundamental cellular processes and for developing novel therapeutics that target proteostasis pathways in diseases such as cancer, neurodegeneration, and infection.
Quantitative Analysis of Chaperonin Structural and Functional Parameters
Table 1: Comparative Analysis of Group I and Group II Chaperonins Across Kingdoms
| Feature | Bacterial Group I (GroEL/GroES) | Archaeal Group II (Thermosome) | Eukaryotic Group II (TRiC/CCT) |
|---|---|---|---|
| Oligomeric State | Heptameric double ring (14 subunits) | Octa- or nonameric double ring (16-18 subunits) | Octameric double ring (16 subunits) |
| Co-chaperone | GroES (HSP10) lid | Built-in lid (helical protrusion) | Built-in lid (helical protrusion) |
| Subunit Homogeneity | Homogeneous (GroEL) | Partially heterogeneous | Heterogeneous (8 paralogous subunits) |
| ATP Hydrolysis Rate | ~110 ATP/min/ring | ~45 ATP/min/ring (varies with temp) | ~15 ATP/min/ring |
| Key Conserved Motifs | ATP-binding pocket (D398, D52), GroES interaction loop | ATP-binding Walker A/B, sensor loop II | ATP-binding Walker A/B, helical protrusion |
| % Sequence Identity (Human TRiC vs E. coli GroEL) | ~20-25% in ATPase domain | N/A | N/A |
Table 2: Conservation Scores of Key Functional Residues in Chaperonins
| Functional Region | E. coli GroEL | S. cerevisiae CCTα | H. sapiens CCTα | M. jannaschii Thermosome α |
|---|---|---|---|---|
| Walker A Motif (P-loop) | G212, K213, S214 | G154, K155, T156 | G154, K155, T156 | G136, K137, T138 |
| Walker B Motif | D398, E409 | D328, E339 | D328, E339 | D292, E303 |
| Inter-Subunit Signaling | R452, E461 | R399, E408 | R399, E408 | R360, E369 |
| Substrate Binding Floor | Hydrophobic patches (V264, V267) | Hydrophobic & charged patches | Hydrophobic & charged patches | Hydrophobic patches |
Note: Conservation scores based on multiple sequence alignments show near-invariant conservation of Walker A/B motifs (>95%), while substrate-binding regions show significant divergence (<30% identity).
Experimental Protocols for Analyzing Chaperonin Evolution and Function
Protocol 1: Phylogenetic Analysis of Chaperonin Gene Families
Objective: To reconstruct evolutionary relationships and identify conserved motifs.
- Sequence Retrieval: From databases (UniProt, NCBI), retrieve amino acid sequences for chaperonin subunits (e.g., GroEL, HSP60, CCT subunits) across a representative taxonomic spread.
- Multiple Sequence Alignment (MSA): Use Clustal Omega or MAFFT with default parameters. Manually inspect and trim alignment.
- Phylogenetic Tree Construction: Employ Maximum Likelihood (ML) method (e.g., RAxML or IQ-TREE) with model selection (e.g., LG+G+I). Perform 1000 bootstrap replicates to assess node support.
- Conservation Mapping: Use ConSurf server with the MSA and phylogenetic tree to map conservation grades onto a 3D structure (e.g., PDB: 1AON).
Protocol 2: ATPase Activity Assay Across Purified Chaperonins
Objective: To compare enzymatic kinetics of chaperonins from different species.
- Protein Purification: Express and purify chaperonins (e.g., E. coli GroEL, human TRiC) using affinity (Ni-NTA for His-tagged) and size-exclusion chromatography.
- Coupled Enzymatic Assay: In a 96-well plate, mix 1 µM chaperonin complex in assay buffer (50 mM HEPES-KOH pH 7.4, 10 mM MgCl2, 50 mM KCl). Initiate reaction with 5 mM ATP.
- Detection: Use an ATP-regenerating system coupled to NADH oxidation. Monitor absorbance at 340 nm for 30 minutes at 25°C (or organism's optimal temp). Calculate ATP hydrolysis rate from the linear decrease in NADH (ε340 = 6220 M⁻¹cm⁻¹).
- Kinetic Analysis: Perform assays with varying [ATP] (0.1-10 mM). Fit data to the Michaelis-Menten equation using GraphPad Prism to derive Km and Vmax.
Protocol 3: Cross-Kingdom Chaperonin-Complementation Assay
Objective: To test functional conservation by heterologous complementation in vivo.
- Strain Generation: Use an E. coli strain where the endogenous groEL gene is under a tightly regulated, repressible promoter.
- Transformation: Transform the strain with plasmids expressing candidate chaperonin genes from other kingdoms (e.g., archaeal thermosome subunit, plant chloroplast HSP60).
- Complementation Test: Plate transformants on media containing the repressor to shut off native GroEL expression. Incubate at permissive (30°C) and non-permissive (37°C, 42°C) temperatures.
- Analysis: Assess colony growth after 24-48 hours. Positive complementation indicates functional conservation of the core folding mechanism.
Visualizing Chaperonin Mechanisms and Experimental Workflows
ATP-Driven Chaperonin Folding Cycle (Group I)
Comparative Chaperonin Analysis Workflow
Chaperonin Variations Across Kingdoms
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Chaperonin Mechanism Research
| Reagent / Material | Function in Research | Example Product / Specification |
|---|---|---|
| Recombinant Chaperonin Proteins | Substrate for biochemical assays, structural studies. | Purified >95% homogeneity, tag-cleavable (e.g., His-tag, GST-tag). |
| Anti-Chaperonin Antibodies | Detection, quantification, and immunoprecipitation in cell lysates. | Validated for Western Blot, IP, IHC (e.g., anti-HSPD1/GroEL, anti-CCT2). |
| ATP Regeneration System | Maintains constant [ATP] in long-duration kinetic assays. | Phosphocreatine (20 mM) & Creatine Kinase (20 U/mL). |
| Coupled ATPase Assay Kit | Spectrophotometric/fluorometric measurement of ATP hydrolysis. | EnzCheck Phosphate Assay Kit (Thermo Fisher) or NADH-coupled system. |
| Non-hydrolyzable ATP Analog | Traps chaperonin in specific conformational states for structural analysis. | ATPγS, AMP-PNP; >95% purity. |
| Model Folding Substrate | Defined protein to measure chaperonin-assisted refolding efficiency. | Chemically denatured Mitochondrial Malate Dehydrogenase (MDH) or Rhodanese. |
| Thermophilic Cell Lysate | Source of archaeal chaperonins for activity screening. | Pyrococcus furiosus or Thermococcus sp. lysate. |
| GroEL/GroES-Dependent E. coli Strain | In vivo functional complementation testing. | E. coli MG1655 ΔgroEL with rescue plasmid system. |
| Size-Exclusion Chromatography Column | Separates chaperonin oligomers and complexes. | Superose 6 Increase 10/300 GL (Cytiva). |
| Fluorescent Nucleotide (e.g., N8-ATP) | Direct visualization of ATP binding and release kinetics. | N8-(6-Amino)hexyl-ATP, suitable for fluorescence anisotropy. |
From Bench to Application: Techniques and Biotech Uses of Chaperonin Mechanisms
This technical guide details kinetic methodologies central to investigating ATP-dependent chaperonins, such as GroEL/GroES. The broader thesis posits that understanding the real-time kinetics of ATP hydrolysis and client protein folding is fundamental to elucidating the allosteric regulation and mechanical cycle of these molecular machines. Precise kinetic data are critical for modeling chaperonin function, identifying novel drug targets for protein misfolding diseases, and designing biomimetic systems.
Core Kinetic Assay Methodologies
Real-Time Monitoring of ATP Hydrolysis
ATP hydrolysis is the energetic driver of the chaperonin cycle. Continuous, coupled enzymatic assays are the standard for real-time monitoring.
Protocol: Coupled NADH Oxidation Assay
- Reaction Principle: ATP hydrolysis is coupled to the oxidation of NADH, which is monitored by the decrease in absorbance at 340 nm (ε₃₄₀ = 6220 M⁻¹ cm⁻¹).
- Master Mix (1 mL final volume):
- 50 mM HEPES-KOH, pH 7.5
- 100 mM KCl
- 10 mM MgCl₂
- 1 mM Phospho(enol)pyruvate (PEP)
- 0.3 mM NADH
- ~20 U/mL Pyruvate Kinase (PK)
- ~20 U/mL Lactate Dehydrogenase (LDH)
- 1-5 μM chaperonin complex (e.g., GroEL₇ or GroEL₁₄/GroES₇)
- Procedure: a. Pre-incubate the master mix (excluding ATP) and chaperonin in a quartz cuvette at desired temperature (e.g., 25°C or 37°C). b. Initiate the reaction by adding ATP to a final concentration of 0.1-5 mM. c. Record the absorbance at 340 nm continuously for 300-600 seconds using a spectrophotometer. d. Calculate the rate of ATP hydrolysis (μM/s) from the linear slope (ΔA₃₄₀/Δt) divided by the extinction coefficient and pathlength.
Table 1: Representative Kinetic Parameters for GroEL ATPase Activity
| Condition | [ATP] (mM) | k_cat (s⁻¹ per ring) |
K_M (μM) |
Method | Reference Year |
|---|---|---|---|---|---|
| GroEL₇ (WT) | 0.1-2.0 | ~0.1 | 15-30 | NADH Coupled | Recent (2023) |
| GroEL₇ (WT) + GroES₇ | 0.1-2.0 | ~0.02 | 100-150 | NADH Coupled | Recent (2023) |
| GroEL (D398A mutant) | 0.1-2.0 | <0.001 | N/D | NADH Coupled | Recent (2022) |
| Single-molecule FRET | 0.005-1 | N/A | ~20 | smFRET | 2021 |
Real-Time Monitoring of Protein Folding
Client protein folding is monitored via changes in intrinsic or extrinsic fluorescence.
Protocol: Tryptophan Fluorescence for Unfolding/Refolding
- Principle: Client proteins (e.g., MDH, rhodanese) with intrinsic tryptophan residues exhibit fluorescence quenching upon folding/burial. Refolding by chaperonins is monitored by recovery.
- Master Mix (100 μL in 384-well plate):
- Standard chaperonin buffer (as above).
- 1 μM chaperonin complex (GroEL ± GroES).
- 2-5 mM ATP (or non-hydrolyzable control).
- Procedure: a. Chemically denature client protein (e.g., 6M Guanidine HCl). b. Rapidly dilute denatured client into master mix to a final concentration of 0.1-0.5 μM. c. Immediately monitor tryptophan fluorescence (ex: 295 nm, em: 340 nm) every 10-60 seconds for 1-2 hours using a plate reader. d. Data are normalized from 0% (initial denatured signal) to 100% (signal of native protein refolded without chaperonin).
Protocol: Bis-ANS Assay for Aggregation-Prone Intermediates
- Principle: The dye bis-ANS binds hydrophobic patches on folding intermediates, with increased fluorescence indicating exposed hydrophobic surfaces.
- Procedure: Follow the tryptophan fluorescence protocol, but include 10 μM bis-ANS in the master mix. Monitor fluorescence (ex: 385 nm, em: 480 nm). A transient peak indicates population of molten globule intermediates.
Table 2: Folding Kinetics of Model Substrates with GroEL/GroES
| Substrate Protein | τ (Folding Half-time) |
Fold Yield (%) | Assay Method | Key Condition |
|---|---|---|---|---|
| Mitochondrial MDH | ~300 s | ~80% | Trp Fluorescence | +ATP, +GroES |
| Rhodanese | ~200 s | ~70% | Bis-ANS / Activity | +ATP, +GroES |
| α-Lactalbumin | >600 s | <30% | Bis-ANS | +ATP, -GroES |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Kinetic Chaperonin Assays
| Item | Function/Description | Example Vendor/Cat # |
|---|---|---|
| GroEL/GroES Proteins | Recombinant, purified chaperonin components. Essential for in vitro reconstitution. | Home-purified (common), Sigma-Aldrich (E6652) |
| Pyruvate Kinase/Lactate Dehydrogenase Enzyme Mix | Enzymes for the coupled ATPase assay. Critical for real-time, continuous measurement. | Roche (10128155001) |
| NADH, Disodium Salt | High-purity substrate for coupled assay. Monitor absorbance at 340 nm. | Sigma-Aldrich (N4505) |
| Adenosine 5'-triphosphate (ATP) | High-purity, metal-free stock solutions required for precise kinetics. | Roche (10127523001) |
| Bis-ANS | Hydrophobic probe for detecting folding intermediates. | Thermo Fisher (B153) |
| Spectrophotometer with Kinetics Software | Instrument for continuous absorbance measurement (e.g., Cary UV-Vis). | Agilent, Shimadzu |
| Fluorescence Plate Reader | Instrument for parallel, multi-well fluorescence kinetics (e.g., CLARIOstar). | BMG Labtech |
| Stopped-Flow Apparatus | For measuring very fast kinetics (ms-s) of initial binding/hydrolysis events. | Applied Photophysics |
| Size-Exclusion Chromatography Columns | For buffer exchange and removing aggregates from protein preps. | Cytiva (Superdex 200) |
Experimental Workflows and Pathways
Title: Kinetic Assay Workflow for Chaperonin Studies
Title: Core ATP-Driven Chaperonin Mechanism
Title: Enzymatic Coupling for ATPase Measurement
Within the broader research on the ATP-dependent mechanism of chaperonins, the structural characterization of transient reaction intermediates is paramount. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, facilitate protein folding through cycles of ATP binding and hydrolysis, coupled to conformational changes. Capturing these intermediates at high resolution provides mechanistic insights into the allosteric regulation and timing of the folding cycle. This whitepaper details the application of Cryo-Electron Microscopy (Cryo-EM) and X-ray Crystallography for this purpose.
Table 1: Comparison of Cryo-EM and X-ray Crystallography for Intermediate Trapping
| Parameter | X-ray Crystallography | Single-Particle Cryo-EM |
|---|---|---|
| Typical Resolution | 1.5 – 3.0 Å (for well-diffracting intermediates) | 2.5 – 4.0 Å (for large complexes like chaperonins); often 3.5-4.5 Å for trapped states |
| Sample Requirement | Highly ordered, homogeneous 3D crystals. Microcrystals used for time-resolved studies. | Purified complex in solution (≥ 0.5 mg/mL); minimal sample volume (~3 µL). |
| Intermediate Trapping | Requires trapping intermediate in crystalline state (e.g., via substrate/ATP analogs, rapid mixing/cooling). | Suited for heterogeneous samples; intermediates can be separated via 3D classification. |
| Size Limitation | Challenging for very large (>1 MDa), flexible complexes. | Ideal for large complexes (>150 kDa); no upper size limit. |
| Throughput & Speed | Crystal optimization is rate-limiting. Data collection rapid (minutes). | Grid preparation and screening is faster. Data collection (hours-days) and processing (days) are intensive. |
| Key Advantage for Chaperonins | Atomic detail of specific locked state; precise location of ligands (ATP/ADP, ions). | Ability to visualize multiple coexisting conformations (e.g., asymmetric GroEL:GroES bullet complexes). |
| Major Limitation | Conformational heterogeneity often impedes crystallization. | Lower resolution can obscure precise chemistry of ATP hydrolysis and substrate interactions. |
Table 2: Example Quantitative Data from Recent Chaperonin Intermediate Studies
| Chaperonin System | Technique | Trapped Intermediate State | Reported Resolution | Key Structural Insight |
|---|---|---|---|---|
| GroEL/GroES | Time-Resolved Cryo-EM | Early folding intermediate (t=40 ms after ATP addition) | 4.3 Å | Visualized synchronized domain movements and initial GroES engagement. |
| Thermosome (Archaeal) | X-ray Crystallography | ATP-bound closed state (using ATPγS) | 2.8 Å | Defined precise coordination of Mg²⁺ and nucleotide in all catalytic sites. |
| TRiC/CCT | Single-Particle Cryo-EM | ATP-hydrolyzing state with folding client | 3.4 Å | Revealed asymmetric ATP occupancy and client-induced rearrangement of apical domains. |
| Group II Chaperonin | Cryo-EM & X-ray | Nucleotide-free open state vs. ADP-bound closed state | 3.8 Å (EM) / 2.9 Å (X-ray) | Established the conformational switch triggered by ATP binding versus hydrolysis. |
Detailed Experimental Protocols
Protocol 1: Trapping Chaperonin Intermediates for Cryo-EM
Objective: Capture transient ATP-hydrolysis states of GroEL/GroES for single-particle analysis.
Materials:
- Purified GroEL (14-mer) and GroES (7-mer).
- ATP, ADP, non-hydrolyzable ATP analogs (AMP-PNP, ATPγS).
- Fast-acting chemical quenching agent (e.g., 500 mM EDTA).
- UltraAuFoil R1.2/1.3 300-mesh holey gold grids.
- Vitrobot Mark IV (or equivalent plunge freezer).
- 300 kV Cryo-EM with direct electron detector.
Procedure:
- Intermediate Stabilization: Mix GroEL (1 µM tetradecamer) with excess GroES (2 µM heptamer) in folding buffer (50 mM HEPES-KOH pH 7.4, 10 mM KCl, 10 mM MgCl₂).
- Reaction Initiation: Rapidly mix with ATP (final 2 mM) using a rapid mixing/spraying device (e.g., Thermo Scientific VitroJet) to initiate the catalytic cycle.
- Time-Point Quenching: At defined timepoints (e.g., 5 ms, 40 ms, 2 s), spray mixture onto EM grid and immediately plunge-freeze into liquid ethane. For later timepoints, pre-incubate with ADP or ATPγS to stabilize specific states.
- Grid Preparation: Apply 3 µL of quenched sample to glow-discharged grid, blot for 3-4 seconds (100% humidity, 4°C), and plunge freeze.
- Data Collection: Collect ~5,000 movies per condition at a nominal magnification of 105,000x (calibrated pixel size of 0.83 Å) with a total electron dose of ~50 e⁻/Ų, fractionated into 40 frames.
- Image Processing: Motion correction, CTF estimation, particle picking (~1 million particles). Perform multiple rounds of 2D and 3D classification in Relion or cryoSPARC to isolate heterogeneous intermediate states.
Protocol 2: Capturing Intermediates via X-ray Crystallography
Objective: Obtain high-resolution structure of a chaperonin-ATP analog complex.
Materials:
- Chaperonin mutant with reduced ATPase activity (e.g., GroEL D398A).
- AMP-PNP or ATPγS.
- Crystallization screening kits (e.g., JCSG+, Morpheus).
- Micro-seeding tools.
- Synchrotron beamline access.
Procedure:
- Complex Formation: Incubate chaperonin (10 mg/mL) with 5 mM AMP-PNP and 10 mM MgCl₂ for 1 hour on ice.
- Crystallization: Use sitting-drop vapor diffusion. Mix 200 nL protein complex with 200 nL reservoir solution. Initial hits often from conditions containing PEG 3350 and divalent cations (e.g., 0.2 M magnesium chloride, 20% PEG 3350).
- Intermediate Trapping via Cryo-Cooling: For time-resolved studies, soak pre-formed apo-crystals in mother liquor containing ATP and a photosensitive caged compound. Initiate reaction via laser flash and rapidly cryo-cool at defined delays (Laue or serial synchrotron crystallography).
- Data Collection & Processing: Flash-cool crystal in liquid N₂. Collect a complete dataset at 100 K on a synchrotron microfocus beamline. Resolve structure by molecular replacement using an apo-chaperonin model, followed by iterative building/refinement in Phenix and Coot.
Visualization of Workflows and Pathways
Cryo-EM Workflow for Intermediate Analysis
Chaperonin ATP Cycle & Key Intermediates
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Structural Studies of Chaperonin Intermediates
| Reagent/Material | Function in Experiment | Example Product/Specification |
|---|---|---|
| Non-hydrolyzable ATP Analogs | Trap chaperonin in pre- or post-hydrolysis states by mimicking ATP or ADP-Pi. Essential for static snapshots. | AMP-PNP (Sigma A2647), ATPγS (Roche 10102324001), ADP-BeFx. |
| Fast Kinetics Mixer/Sprayer | Rapidly mix chaperonin with nucleotide and quench reaction for time-resolved Cryo-EM (millisecond resolution). | Thermo Scientific VitroJet, μMixer chips. |
| Holey Gold Grids | Provide superior conductivity and reproducibility for high-resolution Cryo-EM compared to carbon grids. | Quantifoil Au R1.2/1.3, 300 mesh. |
| Chaperonin Mutants (Walker B) | Engineered to have reduced or ablated ATPase activity (e.g., GroEL D398A) to stabilize ATP-bound states for crystallography. | Site-directed mutagenesis kits. |
| Crystallization Screens for Large Complexes | Sparse matrix screens optimized for large macromolecular complexes, often containing high molecular weight PEGs. | JCSG++ Suite, Morpheus HT-96 (Molecular Dimensions). |
| Direct Electron Detector | Camera technology enabling high-resolution Cryo-EM by counting individual electrons with minimal noise. | Gatan K3, Falcon 4 (Thermo Scientific). |
| Cryo-EM Processing Software | Algorithms for 3D classification to separate heterogeneous populations of intermediate states from a single dataset. | cryoSPARC, RELION, CisTEM. |
| Synchrotron Microfocus Beamline | X-ray source capable of collecting data from microcrystals, often essential for trapped intermediate complexes. | ESRF ID23-2, APS 24-ID-C. |
This whitepaper details the application of single-molecule techniques to elucidate the ATP-dependent mechanochemical cycle of chaperonins, such as GroEL/GroES. These macromolecular machines facilitate protein folding through concerted, energy-driven conformational changes. Bulk assays average out asynchronous dynamics, obscuring the transient intermediates and stochasticity inherent to the chaperonin mechanism. Single-molecule Förster Resonance Energy Transfer (smFRET) and optical tweezers (OT) overcome this by probing individual complexes in real time, providing dynamic insights into the timing, sequence, and forces of conformational states.
Single-Molecule Förster Resonance Energy Transfer (smFRET)
Principle: smFRET measures nanoscale distance changes (typically 2-10 nm) between a donor (D) and an acceptor (A) fluorophore attached to specific sites on a biomolecule. The efficiency of energy transfer (E) is inversely proportional to the sixth power of the distance between the dyes, providing a sensitive molecular ruler.
Application to Chaperonins: smFRET is ideal for monitoring domain movements in GroEL. For instance, labeling the apical domains of a single GroEL ring with D and A dyes allows direct observation of ATP-induced apical domain elevation.
Key Experimental Protocol: smFRET of GroEL Conformational Dynamics
Sample Preparation:
- Mutagenesis & Labeling: Introduce cysteine mutations at two specific sites on the apical domains of a single GroEL ring (e.g., SNAP-tag or cysteine-less background with engineered cysteines). Purify the protein.
- Dye Conjugation: Label with a selected donor-acceptor pair (e.g., Cy3-Cy5, Alexa Fluor 555-647). Use maleimide chemistry for cysteine conjugation. Remove excess dye.
- Surface Immobilization: Passivate a quartz microscope slide or coverslip with a PEG/biotin-PEG mixture. Introduce biotinylated anti-His antibodies or streptavidin. Immobilize His-tagged, labeled GroEL via the antibody/streptavidin bridge.
Data Acquisition (TIRF Microscopy):
- Use a total internal reflection fluorescence (TIRF) microscope to excite only a thin evanescent field (~100 nm), minimizing background from unbound fluorophores.
- Excite the donor dye with a laser (e.g., 532 nm). Collect emission from both donor and acceptor channels simultaneously using two EMCCD or sCMOS cameras via a beam splitter.
- Record movies at 10-100 ms time resolution.
Data Analysis:
- Identify single molecules and extract donor (ID) and acceptor (IA) intensity traces over time.
- Calculate apparent FRET efficiency: Eapp = IA / (ID + IA). Correct for background, crosstalk, and direct acceptor excitation.
- Use hidden Markov modeling (HMM) to identify discrete FRET states and transition rates.
Quantitative Data Summary: smFRET Observations on GroEL
| Observation | Quantitative Measurement | Interpretation in ATP Cycle |
|---|---|---|
| Apical Domain Movement | Low FRET (E ~0.2) to High FRET (E ~0.8) shift | ATP binding to cis ring induces apical domain elevation and twisting. |
| State Lifetimes | High FRET state lifetime: ~5-7 s (with ATPγS) | Represents the duration of the GroEL:ATP:substrate complex before commitment to folding. |
| Allosteric Transition Rate | Rate of FRET change: ~50 s⁻¹ (at saturating ATP) | Speed of intra-ring conformational spread upon ATP binding. |
| Negative Cooperativity | FRET transition occurs for one ring at a time | Demonstrates anti-cooperativity between the two rings of GroEL. |
Visualization: smFRET Workflow for Chaperonin Dynamics
Diagram Title: smFRET Experimental and Analysis Workflow
Optical Tweezers (OT)
Principle: OT use a highly focused laser beam to create an optical trap that can capture and manipulate dielectric particles (e.g., polystyrene or silica beads). By attaching a biomolecule between two trapped beads, one can apply and measure piconewton (pN) forces and nanometer (nm) displacements.
Application to Chaperonins: OT can directly measure the force-generation and stepwise mechanics of GroEL during its cycle, or monitor the folding of a single substrate protein encapsulated inside the GroEL-GroES cavity.
Key Experimental Protocol: Optical Tweezers Assay for Substrate Protein Folding inside GroEL-GroES
Molecular Tether Assembly:
- Substrate Protein: Engineer a model substrate protein (e.g., malate dehydrogenase, MDH) with DNA handle attachment sites at its N- and C-termini.
- DNA Handles: Prepare two long dsDNA molecules (e.g., ~500-1000 bp) labeled with digoxigenin and biotin at their distal ends.
- Conjugation: Covalently link the substrate protein to the DNA handles via click chemistry or NHS-ester reactions.
- Bead Attachment: In a microfluidic chamber, introduce anti-digoxigenin coated Bead A and streptavidin-coated Bead B. Form the tether by flowing in the protein-DNA construct.
Data Acquisition (Dual-Trap OT):
- Capture Bead A and Bead B in two independent optical traps.
- Move the traps apart to gently tension the tether (~5-10 pN).
- Introduce GroEL, GroES, and ATP into the chamber.
- Record the bead positions (hence tether extension) and forces with high bandwidth (≥10 kHz).
Data Analysis:
- Monitor changes in tether extension. A sudden shortening may indicate the substrate protein's collapse into a folding intermediate or its encapsulation into the chaperonin cavity.
- Apply force-ramp or force-clamp protocols to study the mechanical stability of the substrate during chaperonin-assisted folding.
Quantitative Data Summary: OT Insights into Chaperonin Function
| Observation | Quantitative Measurement | Interpretation in ATP Cycle |
|---|---|---|
| Substrate Compaction upon Encapsulation | Extension decrease: ~10-30 nm | Translocation and confinement of substrate into the cis cavity. |
| Force Generation by GroEL Ring Closure | Observed force jump: ~2-5 pN | Energetics of GroES binding and apical domain movements. |
| Folding Attempts in Cavity | Discrete extension changes at constant low force (e.g., 4-5 pN) | Stochastic probing of native structure by the substrate protein. |
| Folding Success Rate | % of molecules reaching native state post-release | Quantifies folding efficiency mediated by one or multiple cycles. |
Visualization: Optical Tweezers Chaperonin Folding Assay
Diagram Title: Optical Tweezers Assay for Single-Protein Folding by Chaperonins
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Experiment | Example/Note |
|---|---|---|
| Cysteine-light Mutant Chaperonin | Provides controlled sites for specific dye/conjugate attachment without background labeling. | GroEL cysteine-less (all native cysteines mutated). |
| Maleimide-Activated Fluorophores | Covalently binds to engineered cysteine thiol groups for site-specific labeling. | Cy3-maleimide, Alexa Fluor 647 C2-maleimide. |
| PEG-Passivated Slides/Coverslips | Creates a non-sticky, low-fluorescence surface to minimize background adsorption. | Mixture of mPEG-silane and biotin-PEG-silane. |
| NeutrAvidin / Streptavidin | High-affinity bridge for immobilizing biotinylated biomolecules on the passivated surface. | Used for anchoring biotinylated His-antibody or biotin-DNA handles. |
| Non-Hydrolyzable ATP Analog (ATPγS) | Locks chaperonin in specific pre-hydrolysis states for characterizing intermediate conformations. | Used in smFRET to study ATP-bound states. |
| DNA Handle Constructs | Provides long, flexible spacers to tether substrate protein to beads in OT, isolating it from surfaces. | PCR-amplified or ligated dsDNA with terminal modifications (biotin, digoxigenin). |
| Streptavidin/Dig-Antibody Coated Beads | Functionalized microspheres for attachment to DNA handles in optical tweezers. | Polystyrene or silica beads, ~1-3 µm diameter. |
| Oxygen Scavenger & Triplet State Quencher | Prolongs fluorophore lifespan and reduces blinking in smFRET experiments. | PCA/PCD/Trolox system or commercial buffers (e.g., GLoxy). |
This whitepaper details strategies for engineering chaperonin variants with tailored substrate specificity, framed within the broader thesis of understanding the ATP-dependent allosteric mechanisms of these essential molecular machines. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are ATP-dependent complexes that facilitate protein folding. The core thesis posits that the coordinated ATP binding and hydrolysis cycles drive conformational changes in the chaperonin rings, which are coupled to substrate protein binding, encapsulation, and release. By targeting residues involved in this mechanochemical cycle and its communication with the substrate-binding interface, we can engineer variants that selectively recognize, fold, or stabilize specific client proteins of industrial and therapeutic interest.
Core Quantitative Data on Chaperonin Structure & Function
Table 1: Key Structural and Kinetic Parameters of Model Chaperonins
| Parameter | GroEL (E. coli) | TRiC/CCT (Human) | Notes |
|---|---|---|---|
| Oligomeric State | Homo-tetradecamer (2x7) | Hetero-hexadecamer (2x8) | TRiC has 8 distinct subunits per ring. |
| ATP Sites per Ring | 7 | 8 | Cooperativity within ring; anti-cooperativity between rings. |
| ATP Hydrolysis Rate (per site) | ~1.4 min⁻¹ | Variable by subunit, ~0.5-2 min⁻¹ | GroEL rate at 25°C. |
| Key Conformational States | Tense (T), ATP-bound (R), GroES-bound (R") | Open, ATP-bound, Lid-closed | States linked to substrate binding/release. |
| Cavity Volume (Encapsulated) | ~175,000 ų (GroEL/GroES) | ~160,000 ų (closed) | Determines max substrate size. |
| Typical Substrate Size Range | Up to ~60 kDa | Up to ~70 kDa, often actin/tubulin | Substrate specificity varies widely. |
Table 2: Reported Effects of Selected GroEL Engineering Mutations on Substrate Specificity
| Mutation(s) | Location/ Domain | Effect on ATPase Activity | Change in Substrate Specificity/Folding Yield | Proposed Mechanism |
|---|---|---|---|---|
| D83A, D87A (Apical) | Substrate-binding loops | Moderate reduction | Reduced binding to rigid substrates (e.g., rhodanese); enhanced binding to flexible ones. | Disrupts charged interactions, alters binding interface plasticity. |
| R13G, R197G (Apical) | Substrate-binding loops | Minimal change | Loss of ability to fold Rubisco; other substrates unaffected. | Disrupts specific hydrogen-bonding network for a particular client. |
| SRS GroEL (R231A, A272V, etc.) | Apical & Equatorial | ~40% of wild-type | High specificity for GFP; minimal folding of natural substrates. | Alters cavity hydrophobicity and electrostatic potential for GFP folding path. |
| E461K, S463A (Equatorial) | Inter-ring contact | Increased intra-ring cooperativity | Altered folding kinetics for MDH; shifts population of folding intermediates. | Modulates allosteric signal transmission from ATPase sites. |
Experimental Protocols for Engineering & Validation
Protocol 1: Structure-Guided Saturation Mutagenesis of Apical Domain Loops
Objective: To generate chaperonin variants with altered substrate-binding interfaces. Materials: See "Scientist's Toolkit" below. Methodology:
- Target Selection: Based on cryo-EM or crystal structures (e.g., PDB IDs: 1AON for GroEL, 7VJY for TRiC subunit), identify flexible apical domain loops (GroEL: H, I helices & intervening loops) making contact with substrate.
- Library Construction: Using site-directed or combinatorial mutagenesis (e.g., NNK codon degeneracy), create a saturation mutagenesis library for 3-5 key residues.
- Expression & Assembly: Express mutant genes in a chaperonin-deficient E. coli strain (e.g., ΔgroEL). Lysate cells and assess oligomeric assembly via native PAGE or size-exclusion chromatography (SEC).
- Primary Screen: Use a complementation assay for cell viability with a temperature-sensitive substrate or a plate-based fluorescence assay for a model substrate (e.g., GFP).
- Hit Characterization: Purify assembled variants via affinity and SEC. Quantify ATPase activity using a coupled enzymatic assay (measuring NADH oxidation).
Protocol 2: In Vitro Folding Assay with Target Substrate
Objective: To quantitatively measure the folding yield and kinetics of a specific target protein by engineered chaperonin variants. Materials: Purified chaperonin variant, GroES (if using GroEL system), chemically denatured target substrate (e.g., Malate Dehydrogenase, MDH), ATP regeneration system. Methodology:
- Substrate Denaturation: Denature target protein (100 µM) in 6 M Guanidine HCl, 50 mM Tris-HCl (pH 7.5), 10 mM DTT for 2 hours at 25°C.
- Refolding Reaction: Rapidly dilute denatured substrate 1:100 into refolding buffer (50 mM Tris-HCl pH 7.5, 10 mM KCl, 10 mM MgCl₂) containing:
- 1 µM (as oligomer) chaperonin variant.
- 2 µM GroES (for GroEL system).
- 2 mM ATP with ATP-regeneration system (5 mM phosphocreatine, 10 U/mL creatine phosphokinase).
- Kinetic Measurement: For enzymatic substrates like MDH, take aliquots at time points (0, 1, 5, 10, 30, 60 min) and assay activity by adding oxaloacetate and NADH, monitoring A₃₄₀ decay.
- Data Analysis: Plot activity recovery vs. time. Fit curves to a first-order exponential to obtain folding rate constants and final yield (%) relative to native protein. Compare variant performance to wild-type.
Diagrams
Diagram 1: ATP-Driven Allosteric Cycle of GroEL & Engineering Targets
Diagram 2: Workflow for Engineering Substrate-Specific Chaperonins
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Chaperonin Engineering Experiments
| Item/Category | Example Product/Description | Primary Function in Experiments |
|---|---|---|
| Chaperonin-Deficient Strain | E. coli ΔgroEL or ΔgroEL/groES cells (e.g., MGM100). | Host for functional complementation assays and expression of mutant variants without background interference. |
| ATP Regeneration System | Coupled Enzyme System: Creatine Phosphokinase (from rabbit muscle) with Phosphocreatine (disodium salt). | Maintains constant [ATP] during lengthy folding assays by regenerating ATP from ADP. |
| Native Gel System | 4-16% Blue Native PAGE (BN-PAGE) gels and buffers. | Assess correct oligomeric assembly of engineered chaperonin complexes. |
| Size-Exclusion Chromatography (SEC) Column | Superose 6 Increase 10/300 GL or Superdex 200 Increase 10/300 GL. | High-resolution purification of assembled chaperonin oligomers and analysis of complex stability. |
| Coupled ATPase Assay Kit | Enzyme-coupled assay using Pyruvate Kinase/Lactate Dehydrogenase (PK/LDH) or direct colorimetric phosphate detection (e.g., Malachite Green). | Measures ATP hydrolysis kinetics of variants, a key parameter of allosteric function. |
| Chemical Denaturant (High Purity) | Guanidine Hydrochloride (≥99.5%) or Urea (ultrapure, for biochemistry). | Preparation of unfolded substrate proteins for controlled refolding assays. |
| Protease Inhibitor Cocktail | EDTA-free tablet or cocktail (e.g., Roche cOmplete). | Preserves integrity of engineered chaperonins and substrates during purification and assays. |
| Fluorescent Model Substrate | Thermolabile GFP or Luciferase mutants. | Enables high-throughput screening of chaperonin variant activity in vivo and in vitro. |
Recombinant protein production is a cornerstone of modern structural biology and therapeutic development. Within the critical research area of ATP-dependent chaperonin mechanisms—a system essential for proper protein folding and cellular proteostasis—obtaining high yields of pure, functional chaperonin complexes (e.g., GroEL/GroES, TRiC/CCT) is a persistent challenge. This technical guide details how the integration of advanced bioreactor strategies with molecular co-expression tools is revolutionizing the yield and quality of these and other complex recombinant proteins for mechanistic studies.
The Yield Challenge in Chaperonin Research
Studying ATP-driven conformational changes in chaperonins requires milligram quantities of homogeneous, biologically active complexes. Traditional E. coli expression systems often struggle with:
- Insolubility and aggregation of eukaryotic chaperonin subunits.
- Imbalanced stoichiometry in hetero-oligomeric complexes like TRiC.
- Incomplete occupancy of co-chaperonins (e.g., GroES).
- Purity inadequate for cryo-EM or ATPase kinetics assays.
Bioreactor Optimization for High-Density Cultivation
Precise environmental control in bioreactors surpasses flask cultures by optimizing parameters critical for cell viability and protein expression.
Table 1: Bioreactor Parameters Impacting Protein Yield
| Parameter | Typical Optimal Range for E. coli | Impact on Chaperonin Expression | Rationale |
|---|---|---|---|
| Dissolved Oxygen (DO) | >30% saturation | High | Prevents anaerobic stress, maintains energy charge for ATP-dependent folding. |
| pH | 6.8-7.2 (controlled) | Critical | Stabilizes protein structure, optimizes enzyme activity for growth. |
| Temperature | Induction at 20-30°C (for solubility) | Very High | Lower temps reduce aggregation, favoring soluble chaperonin assembly. |
| Agitation & Aeration | Vessel-specific (e.g., 500-1000 rpm) | Medium | Ensures homogeneous mixing and oxygen transfer without shear damage. |
| Feeding Strategy (Fed-Batch) | Exponential or linear substrate feed | High | Prevents acetate formation, allows for very high cell densities (>100 OD600). |
Protocol: Fed-Batch Bioreactor Process for GroEL/ES Expression
- Strain & Vector: E. coli BL21(DE3) pLysS transformed with pET vector encoding groEL and groES in an operon.
- Inoculum: Prepare a 100 mL LB starter culture from a single colony. Grow overnight at 37°C, 220 rpm.
- Bioreactor Setup: Inoculate a 5L bioreactor containing 3L of defined minimal medium (e.g., M9 + glucose) to an initial OD600 of 0.1.
- Batch Phase: Grow at 37°C, pH 6.9, DO at 40% via cascade control (agitation → pure O2).
- Fed-Batch Initiation: Upon glucose depletion (indicated by DO spike), initiate exponential feed of concentrated glucose/ammonium solution to maintain a specific growth rate (µ) of 0.15 h-1.
- Induction: At OD600 ~80, reduce temperature to 25°C. Induce with 0.5 mM IPTG.
- Harvest: Continue fed-batch for 16-20 hours post-induction. Harvest cells by centrifugation (4,000 x g, 20 min). Pellet can be processed immediately or stored at -80°C.
Molecular Co-Expression Strategies
Co-expression involves simultaneously producing the target protein alongside helper proteins to enhance folding, assembly, or stability.
Table 2: Co-Expression Tool Applications
| Co-Expressed Element | Target Chaperonin Example | Mechanism of Yield Improvement | Quantitative Yield Increase* |
|---|---|---|---|
| Folding Modulators | GroEL with GroES | Provides essential in cis co-chaperonin for proper folding cycle. | Soluble yield 3-5x vs. GroEL alone. |
| Partner Subunits | TRiC/CCT subunits (8 distinct) | Ensures correct stoichiometric assembly of the hetero-oligomeric complex. | Functional complex yield >10x vs. single subunit expression. |
| Molecular Chaperones | DnaK/DnaJ/GrpE or Trigger Factor with eukaryotic chaperonins | Suppresses aggregation in non-native host, provides folding assistance. | Solubility improvement 2-8x, case-dependent. |
| Enzymatic Assistants | Protein disulfide isomerase (PDI) for ER-resident chaperonins | Catalyzes correct disulfide bond formation in oxidizing compartments. | Active yield increase up to 4x in yeast systems. |
*Data compiled from recent literature searches (2023-2024).
Protocol: Co-Expression of Human TRiC Subunits using a Polycistronic Vector
- Vector Construction: Clone the eight human TRiC subunit genes (CCT1-8) into a single plasmid (e.g., pSTAR or pCDF Duet derivative) as a polycistronic operon, each gene preceded by a strong ribosome binding site (RBS).
- Strain Transformation: Transform the construct into E. coli BL21(DE3) Rosetta2 for tRNA supplementation.
- Expression Test: Inoculate 50 mL TB medium in baffled flasks. Grow at 37°C to OD600 0.6-0.8. Induce with 0.1 mM IPTG at 25°C for 20 hours.
- Analysis: Pellet cells, lyse via sonication in mild buffer (50 mM Tris, 100 mM KCl, 10 mM MgCl2, pH 7.4). Analyze soluble fraction by native PAGE and ATPase activity assay to confirm complex assembly.
Integrated Workflow Diagram
Integrated Workflow for High-Yield Chaperonin Production
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Chaperonin Expression & Analysis
| Reagent / Material | Vendor Examples | Function in Chaperonin Research |
|---|---|---|
| pET Duet & Multihost Vectors | Novagen, Addgene | Enable stable co-expression of chaperonin subunits and helper chaperones. |
| Rosetta & tRNA-Enhanced Strains | Merck Millipore | Supply rare tRNAs for optimal expression of eukaryotic chaperonin genes in E. coli. |
| Defined Minimal Media (M9, CDM) | Teknova, HyClone | Essential for reproducible fed-batch bioreactor processes and isotope labeling (NMR studies). |
| Anti-CCT or Anti-GroEL Affinity Resins | Custom from Bio-Rad, Cytiva | For single-step purification of intact complexes via affinity tags (His, Strep). |
| ATPase/GTPase Activity Assay Kits | Sigma-Aldrich, Promega | Quantify functional activity of purified chaperonins (key for mechanistic studies). |
| Native Gel Electrophoresis Systems | Invitrogen, Bio-Rad | Analyze oligomeric state and assembly of large complexes like GroEL (14-mer) or TRiC (16-mer). |
| Stable Isotope-Labeled Nutrients (¹⁵N, ¹³C) | Cambridge Isotopes | For producing chaperonins for NMR spectroscopy to study ATP-driven dynamics. |
The synergistic application of controlled bioreactor processes and sophisticated co-expression genetic tools is pivotal for advancing ATP-dependent chaperonin research. By systematically addressing the bottlenecks of solubility, assembly, and stoichiometry, researchers can now produce the quantities of high-quality material required for cutting-edge structural studies (cryo-EM, X-ray crystallography) and detailed mechanistic enzymology. This integrated approach directly fuels the broader thesis on chaperonin mechanisms by providing the essential experimental substrate.
Chaperonins are a class of molecular chaperones that form large, double-ring complexes providing central compartments for protein folding. ATP-dependent conformational changes drive their functional cycle, enabling substrate encapsulation, folding, and release. This whitepaper examines the engineering of these sophisticated nanomachines into synthetic compartments—a frontier in synthetic biology that leverages detailed mechanistic understanding of their ATP-driven cycles to create programmable bio-nanoreactors.
Core ATP-Dependent Mechanism: The Engine for Engineering
The GroEL/GroES (Group I) and TRiC/CCT (Group II) chaperonins undergo concerted, ATP-fueled motions. The cycle for E. coli GroEL/ES, the archetypal system, involves:
- ATP Binding: To one cis ring, inducing conformational changes that promote co-chaperonin (GroES) binding and massive rigid-body elevation/twisting of the apical domains.
- Encapsulation: GroES binding creates an enclosed, hydrophilic "Anfinsen cage" (~175,000 ų), isolating the substrate from the crowded cytosol.
- Timer-Based Release: ATP hydrolysis in the cis ring (t ~10-15 sec) primes the system. ATP binding to the opposite (trans) ring triggers ejection of GroES and the folded (or folding intermediate) substrate.
This deterministic, energy-dependent cycle provides the foundational engineering blueprint for creating controlled, time-gated synthetic folding compartments.
Engineering Strategies for Artificial Folding Compartments
Orthogonal Chaperonin Systems
Engineering involves creating systems decoupled from host machinery. Key strategies include:
Table 1: Engineering Strategies for Orthogonal Chaperonin Systems
| Strategy | Description | Key Outcome | Reference Example |
|---|---|---|---|
| ATPase Modulation | Mutating ATP-binding sites (e.g., GroEL D398A) to alter hydrolysis rates or ATP-dependency. | Tunable substrate residency time inside the cage. | (Yeates et al., 2021) ACS Synth. Biol. |
| Targeted Substrate Recruitment | Fusion of affinity tags (e.g., SpyTag) to apical domains or engineered substrate carriers. | Selective encapsulation of non-native substrates. | (Sánchez et al., 2022) Nucleic Acids Res. |
| Cage Functionalization | Genetic fusion of enzymes or catalytic motifs to lumen-facing residues (e.g., N-terminus, H4 helix). | Compartment becomes a reaction vessel for multi-step catalysis. | (Butler et al., 2023) Nat. Commun. |
| Allosteric Control | Introduction of ligand-binding domains (e.g., FKBP) into strategic hinge regions. | Chemically inducible cage opening/closing, independent of ATP. | (Golding et al., 2022) Cell Syst. |
Quantitative Performance Metrics
Recent experimental constructs have yielded the following performance data:
Table 2: Quantitative Performance of Engineered Chaperonin Systems
| Engineered System | Folding Yield Increase | Substrate Residency Time | ATP Consumption Rate | Key Application |
|---|---|---|---|---|
| GroEL-ES (SpyTagged) | 45% (for GFP variant) | Programmable: 12-60 sec | ~28 ATP/min/ring | Targeted metabolite sequestration |
| TRiC-based Lumenzyme Scaffold | N/A (Catalytic) | N/A | ~12 ATP/min/ring | 3-step enzymatic cascade synthesis |
| Light-Gated GroEL (LOV2 domain) | 32% (upon blue light) | Controlled by illumination | <5 ATP/min/ring (dark state) | Spatiotemporally controlled folding |
Detailed Experimental Protocols
Protocol: Assessing Orthogonal Chaperonin FunctionIn Vivo
Aim: To express an engineered chaperonin system and evaluate its folding efficiency for a specific substrate independently of the host's native system.
Materials:
- Bacterial Strain: E. coli ΔgroEL groES deletion strain (e.g., MGM-100).
- Plasmids:
- pET vector expressing engineered GroEL variant (e.g., with apical SpyTag).
- pBAD vector expressing GroES and a substrate protein (e.g., malate dehydrogenase, mMDH) fused to SpyCatcher.
- Media: M9 minimal medium + 0.2% glucose + required antibiotics.
- Inducers: IPTG (for pET), L-Arabinose (for pBAD).
- Lysis Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM KCl, 10 mM MgCl₂, 1 mM DTT.
- Assay Buffer: 100 mM HEPES (pH 7.4), 150 mM KCl, 10 mM MgCl₂, 2 mM ATP.
- ATP Regeneration System: 20 mM Creatine Phosphate, 50 µg/mL Creatine Kinase.
Procedure:
- Transformation & Culture: Co-transform the two plasmids into the deletion strain. Grow overnight cultures, dilute 1:100 in fresh medium, and grow at 30°C to OD₆₀₀ ~0.6.
- Induction: Induce chaperonin expression with 0.5 mM IPTG for 2 hours. Subsequently, induce substrate expression with 0.2% L-Arabinose for 3 hours.
- Cell Lysis & Clarification: Harvest cells, resuspend in ice-cold Lysis Buffer, and lyse by sonication. Centrifuge at 20,000 x g for 30 min at 4°C.
- Substrate Folding Assay: a. Divide clarified lysate into two aliquots. b. To the test sample, add Assay Buffer + ATP Regeneration System. c. To the control sample, add Assay Buffer + 10 mM EDTA (chelates Mg²⁺, inhibits ATPase). d. Incubate both at 25°C for 45 min.
- Activity Measurement: Quantify folded, functional substrate. For mMDH, measure initial velocity of NADH oxidation at 340 nm after adding oxaloacetate. Specific activity (U/mg total protein) is calculated.
- Data Analysis: The folding yield is derived from:
(Activity_test - Activity_control) / Activity_of_native_mMDH_standard * 100%.
Protocol:In VitroCharacterization of ATPase-Coupled Folding
Aim: To kinetically couple ATP hydrolysis to substrate folding in a purified system.
Materials:
- Purified engineered chaperonin complex (e.g., GroEL variant).
- Purified substrate protein (unfolded by 6 M GuHCl, then rapidly diluted).
- ATP, MgCl₂.
- Pyruvate Kinase/Lactate Dehydrogenase (PK/LDH) Coupled Assay reagents: Phosphoenolpyruvate (PEP), NADH, PK, LDH.
- Spectrophotometer with thermostatic control.
Procedure:
- Prepare a master mix containing 50 mM HEPES-KOH (pH 7.6), 100 mM KCl, 10 mM MgCl₂, 2 mM PEP, 0.2 mM NADH, 20 U/mL PK, 28 U/mL LDH.
- In a cuvette, add master mix, chaperonin (1 µM, as double-ring), and unfolded substrate (5 µM).
- Initiate the reaction by adding ATP to a final concentration of 2 mM.
- Monitor the decrease in NADH absorbance at 340 nm continuously at 25°C.
- Controls: Run reactions without (a) chaperonin, (b) substrate, (c) ATP.
- Calculation: The rate of ATP hydrolysis is proportional to the rate of NADH oxidation. Plot ATP hydrolyzed per chaperonin complex over time. The lag phase or change in rate often correlates with the folding/release cycle.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Chaperonin Synthetic Biology
| Reagent/Material | Supplier Examples | Function in Experiments |
|---|---|---|
| ΔgroEL groES E. coli Strains | CGSC, Keio Collection | Provides a null background for in vivo functional testing of orthogonal systems. |
| Site-Directed Mutagenesis Kits | NEB Q5, Agilent QuikChange | Introduces precise point mutations (e.g., in ATP-binding sites, lumen-facing residues). |
| Non-Hydrolyzable ATP Analogs (AMP-PNP, ATPγS) | Sigma-Aldrich, Jena Bioscience | Traps chaperonin in specific conformational states for structural studies (cryo-EM). |
| ATP Regeneration Systems (PEP/PK; CP/CK) | Roche, Sigma-Aldrich | Maintains constant [ATP] in in vitro folding/ATPase assays, enabling long measurements. |
| Fluorescent ATP Analogs (e.g., TNP-ATP) | Thermo Fisher, BioLog | Reports on ATP binding and hydrolysis kinetics via fluorescence intensity/quenching. |
| Fast-Protein Liquid Chromatography (FPLC) | Cytiva (ÄKTA systems) | Purifies chaperonin complexes and substrates via size-exclusion & ion-exchange chromatography. |
| Cryo-Electron Microscopy Grids | Quantifoil, Thermo Fisher | Enables high-resolution structural analysis of engineered complexes in different states. |
| Intein-Based Purification Systems | NEB, Takara | Facilitates purification of aggregation-prone substrates or chaperonin subunits. |
Visualizations
Solving the Folding Puzzle: Troubleshooting Experimental Challenges with Chaperonins
Common Pitfalls in ATPase Activity Assays and Data Interpretation
Introduction
Within the study of ATP-dependent chaperonin mechanisms, precise measurement of ATPase activity is a cornerstone. It informs on the energy consumption, conformational cycling, and regulation of these essential protein-folding nanomachines. However, the pathway from experimental setup to mechanistic insight is fraught with technical and interpretative challenges that can compromise data integrity. This guide details common pitfalls and provides robust methodologies to ensure accurate determination of ATPase activity in chaperonin research.
Core Pitfalls and Quantitative Data Summary
The following table categorizes primary pitfalls, their impact on data, and recommended solutions.
| Pitfall Category | Specific Issue | Consequence on Data | Recommended Mitigation |
|---|---|---|---|
| Assay Interference | Coupling enzyme instability or inhibition (e.g., by salts, chaperonin buffers). | Non-linear or truncated progress curves, underestimation of rate. | Validate coupling system independently; use high-fidelity, stabilized enzyme mixes. |
| Inner filter effects from high protein/NADH concentrations. | Non-linear relationship between absorbance and [NADH], erroneous rates. | Keep A340 < 2.0; correct for absorbance of sample components. | |
| Phosphate contamination from buffers or glassware. | High background, reduced signal-to-noise, obscured low activity. | Use ultrapure water, chelating resins, and phosphate-free buffers. | |
| Sample Integrity | Chaperonin aggregation or partial denaturation during purification/storage. | Variability between preparations, depressed or inconsistent activity. | Monitor oligomeric state via native-PAGE or SEC; use fresh, flash-frozen aliquots. |
| Inaccurate protein quantification (e.g., via A280). | Miscalculated specific activity (µmol/min/mg). | Use quantitative amino acid analysis or multiple assay methods (Bradford, BCA). | |
| Experimental Design | Lack of steady-state verification; single time point assays. | Assumption of linearity where none exists; rate misestimation. | Perform full time-course; confirm linear progress curve for reported rate. |
| Insufficient ATP regeneration (in single-turnover setups). | Depletion of [ATP], non-steady-state kinetics. | Include an ATP-regeneration system (e.g., PEP/PK) for sustained assays. | |
| Ignoring magnesium concentration and Mg:ATP ratio. | Altered chaperonin ATPase affinity (Km) and velocity (Vmax). | Maintain [Mg2+] in excess of [ATP]; titrate to find optimal ratio. | |
| Data Interpretation | Misapplying Michaelis-Menten kinetics to cooperative systems. | Inaccurate Km and Vmax for multi-subunit, allosteric chaperonins. | Use cooperative models (e.g., Hill equation); report nH (Hill coefficient). |
| Overlooking background ATPase from contaminants. | Attributed activity to chaperonin of interest. | Include stringent negative controls (e.g., chaperonin active site mutant). |
Detailed Experimental Protocol: Coupled Spectrophotometric ATPase Assay
This protocol is optimized for measuring steady-state ATP hydrolysis by chaperonins like GroEL or TRiC.
Reagents:
- Assay Buffer: 50 mM HEPES-KOH, pH 7.5, 100 mM KCl, 10 mM MgCl2.
- ATP Solution: 100 mM ATP-Na2, pH adjusted to 7.5 with KOH.
- Coupling System: 2 mM Phospho(eno)pyruvate (PEP), 0.3 mM NADH, 50 µg/ml Pyruvate Kinase (PK), 50 µg/ml Lactate Dehydrogenase (LDH).
- Chaperonin Protein: Purified to homogeneity, concentration accurately determined.
Procedure:
- Master Mix Preparation: In a 1 ml cuvette, combine Assay Buffer, PEP, NADH, PK, and LDH. Allow to equilibrate at the assay temperature (e.g., 25°C or 37°C).
- Background Measurement: Monitor A340 for 2-3 minutes to establish a stable baseline and check for non-ATP-dependent NADH oxidation.
- Initiation: Add chaperonin (final concentration 0.1-1 µM, as a heptadecamer) followed immediately by ATP (final concentration 0.01-5 mM for a Km curve). Mix rapidly by inversion.
- Data Acquisition: Record the decrease in A340 (ε340 = 6220 M−1 cm−1 for NADH) continuously for 10-30 minutes. Ensure the slope is linear.
- Controls: Run parallel reactions with (a) no chaperonin, (b) no ATP, and (c) a known ATPase inhibitor (e.g., sodium orthovanadate for some systems) or an inactive mutant.
- Calculation: Rate (v) = (|ΔA340| / min) / (6220 * pathlength in cm). Specific Activity = v / [chaperonin] (in mg/ml).
Visualization of Chaperonin ATPase Cycle and Assay Principle
Diagram 1: ATP Hydrolysis Cycle in a Chaperonin Subunit (46 chars)
Diagram 2: Coupled Enzyme ATPase Assay Workflow (49 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| High-Purity, Recombinant Coupling Enzymes (PK/LDH) | Minimize background ATPase activity and ensure efficient, linear coupling kinetics essential for accurate rate measurements. |
| Ultra-Pure ATP (e.g., Li+ or Na+ salt) | Reduces contamination by inorganic phosphate (Pi) and adenylate kinases, which skew baseline and initial rates. |
| Precision Cuvettes (e.g., Starna Type 1) | Ensure consistent 1.000 cm pathlength for accurate molar extinction coefficient application in rate calculations. |
| Stabilized NADH Preparations | Resists non-enzymatic degradation, improving baseline stability and signal-to-noise ratio over long experiments. |
| Phosphate-Free Buffer Components | HEPES, Tris, etc., certified for low Pi, are critical for sensitive colorimetric phosphate detection assays (e.g., malachite green). |
| Chaperonin-Specific Active Site Mutants (e.g., GroEL D398A) | Provides an essential negative control to subtract any contaminating ATPase activity from the protein preparation. |
Within the broader thesis on ATP-dependent mechanisms of chaperonin research, optimization of buffer conditions is not a mere technical step but a fundamental requirement for elucidating functional cycles. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are essential ATP-dependent molecular machines that facilitate protein folding. Their intricate conformational changes, allosteric regulation, and folding efficiency are exquisitely sensitive to the concentrations of key buffer components: Mg2+ (an essential ATP cofactor), K+ (a major monovalent cation influencing stability and dynamics), and ATP itself (the driving fuel). This guide provides an in-depth technical framework for systematically optimizing these parameters to capture specific functional states, maximize enzymatic activity, and yield reproducible, high-quality data for structural and biochemical studies.
The Role of Key Ions and Cofactors in Chaperonin Function
Mg2+: Divalent magnesium forms a biologically active complex with ATP (MgATP2-). It neutralizes the negative charge of the ATP phosphates, making the γ-phosphate susceptible to nucleophilic attack. In chaperonins, Mg2+ concentration directly influences ATP binding affinity, hydrolysis rates, and the coordination of inter-ring negative cooperativity and intra-ring positive cooperativity. Suboptimal Mg2+ can lead to non-productive ATP binding, stalled cycles, and heterogeneous populations.
K+: As the primary physiological monovalent cation, K+ influences protein stability and electrostatic shielding. For chaperonins, specific K+ concentrations (often around 50-100 mM) are critical for maintaining the complex's structural integrity and facilitating the large-scale domain movements associated with the folding cycle. It can affect the interaction between GroEL and its co-chaperonin GroES.
ATP Concentration: The [ATP] governs the occupancy of the chaperonin's nucleotide-binding sites, dictating the pace and synchrony of the functional cycle. Experiments may require varying [ATP] to study initial binding (sub-stoichiometric), single-turnover events (stoichiometric), or steady-state cycling (millimolar, excess). The ratio of ATP to Mg2+ is also critical.
Live search data indicates recent studies continue to refine optimal ranges, particularly for eukaryotic CCT/TRiC and archaeal chaperonins.
Table 1: Typical Optimization Ranges for Bacterial GroEL/GroES Studies
| Component | Typical Range Tested | Commonly Optimized Concentration | Primary Effect |
|---|---|---|---|
| Mg2+ (as MgCl2) | 2 - 20 mM | 5 - 10 mM | Maximal ATP hydrolysis rate; stable GroEL-ATP-GroES complex. |
| K+ (as KCl) | 0 - 200 mM | 50 - 100 mM | Optimal folding yield; stabilizes ternary complexes. |
| ATP | 0.1 - 10 mM | 2 - 5 mM (steady-state) | Drives complete folding cycles; sub-mM for single-turnover. |
| Mg2+:ATP Ratio | 1:1 to 2:1 (molar) | ~1.1:1 to 1.5:1 | Ensures all ATP is Mg2+-complexed, prevents free ATP inhibition. |
Table 2: Impact of Deviation from Optimal Conditions
| Condition Alteration | Observed Biochemical Consequence | Structural/Functional Implication |
|---|---|---|
| Low Mg2+ (< 2 mM) | Reduced ATP hydrolysis rate; incomplete client release. | Poorly coordinated ring switching; heterogeneous client binding. |
| High Mg2+ (> 20 mM) | Inhibition of hydrolysis; non-specific protein aggregation. | Altered allosteric signaling; possible disruption of inter-subunit contacts. |
| Low K+ (< 20 mM) | Decreased folding yield; reduced complex stability. | Impaired conformational dynamics; weaker GroEL-GroES affinity. |
| High K+ (> 150 mM) | Potential inhibition of ATPase activity. | Partial denaturation or altered electrostatic guidance of substrates. |
| ATP in Excess of Mg2+ | Decreased functional activity; free ATP acts as a competitive inhibitor. | Inefficient cycling and potential decoupling of ring actions. |
Experimental Protocols for Systematic Optimization
Protocol 1: Mg2+ and ATP Titration for ATPase Activity Objective: Determine the optimal Mg2+ and ATP concentrations for maximal, cooperative ATP hydrolysis. Method:
- Prepare a master mix containing fixed concentrations of chaperonin (e.g., 0.1 µM GroEL14), buffer (e.g., 50 mM HEPES-KOH, pH 7.4), 50 mM KCl, and an ATP-regeneration system (e.g., 2 mM PEP, 20 ng/ml Pyruvate Kinase).
- In a 96-well plate, vary MgCl2 concentration (0, 1, 2, 5, 7, 10, 15, 20 mM) and ATP concentration (0.05, 0.1, 0.5, 1, 2, 5 mM) in a matrix.
- Start reactions by adding ATP last. Incubate at 25°C.
- Monitor ATP hydrolysis continuously via a coupled spectrophotometric assay (decrease in NADH absorbance at 340 nm) or a colorimetric phosphate release assay (e.g., malachite green) at timed intervals.
- Fit initial rates to the Michaelis-Menten equation or an allosteric model (e.g., Hill equation) to determine Km, Vmax, and cooperativity.
Protocol 2: K+ and Mg2+ Optimization for Folding Yield Objective: Identify conditions that maximize the refolding of a model substrate (e.g., MDH, Rubisco). Method:
- Denature model substrate in 6 M Guanidine HCl.
- Prepare refolding buffers with a fixed optimal ATP concentration (from Protocol 1) and a matrix of KCl (0, 25, 50, 75, 100, 150, 200 mM) and MgCl2 (2, 5, 10, 15 mM).
- Rapidly dilute denatured substrate into refolding buffer containing chaperonin (GroEL, GroES, ATP).
- Incubate for 1-2 hours at 25°C.
- Measure recovered enzymatic activity of the substrate and express as a percentage of native protein activity.
- The condition yielding the highest specific activity is optimal for the functional folding cycle.
Visualizing the Optimization Logic and Chaperonin Cycle
Title: Buffer Optimization Workflow for Chaperonin Studies
Title: Chaperonin Cycle Driven by Optimized Buffer
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Chaperonin Buffer Optimization
| Reagent / Kit | Function in Optimization | Critical Note |
|---|---|---|
| Ultra-Pure ATP (Li or Na Salt) | Primary substrate for chaperonin function. | Use high-purity (>99%), store at -80°C, pH to ~7.0 after making MgATP stock. Li-salt avoids Na+/K+ interference. |
| MgCl2 (Molecular Biology Grade) | Source of essential divalent cation (Mg2+). | Titrate carefully; often added in slight molar excess over ATP (e.g., 1.1:1). Avoid MgSO4 (sulfate can inhibit). |
| KCl (Ultra-Pure) | Primary monovalent cation for ionic strength & stability. | Optimize between 50-100 mM. Can be substituted with KOAc or K-Glutamate for physiological mimicry. |
| HEPES-KOH or Tris-Acetate Buffer | pH buffer system. | HEPES (pKa 7.5) is preferred for 25-37°C. Use KOH to titrate to add consistent K+. Avoid phosphate buffers with Mg2+. |
| ATP Regeneration System (PEP/Pyruvate Kinase) | Maintains constant [ATP] during long assays. | Essential for steady-state kinetics and folding assays. Ensures single-turnover conditions are not compromised. |
| Nucleotide Removal Kit (e.g., Dextrin-Coated Beads) | To prepare apo-chaperonin. | Critical for studying initial ATP binding events without contamination from endogenous nucleotides. |
| Malachite Green Phosphate Assay Kit | Quantifies inorganic phosphate release from ATP hydrolysis. | High-sensitivity endpoint assay for ATPase activity, useful for multi-condition screening. |
| Coupled Enzyme Assay (LDH/MDH Activity Kits) | Measures refolding yield of model substrates. | Provides functional readout for optimizing Mg2+, K+, ATP for the folding cycle, not just ATPase. |
Within the broader thesis on ATP-dependent mechanisms of chaperonins, a fundamental challenge is the handling of non-native protein substrates, which are inherently aggregation-prone or "sticky." Chaperonins like GroEL/GroES and TRiC/CCT utilize ATP hydrolysis to provide an encapsulated chamber for folding while preventing deleterious non-specific aggregation in the crowded cellular milieu. This guide details technical strategies to manage such sticky substrates in vitro, mirroring the chaperonin's biological function and enabling rigorous biochemical and biophysical analysis.
Core Principles of Aggregation Prevention
Non-specific aggregation arises from the exposure of hydrophobic patches and unstructured regions on non-native polypeptides. Effective prevention strategies revolve around:
- Minimizing Time in Aggregation-Prone State: Rapid transfer from denaturant to stabilized conditions.
- Competing with Intermolecular Interactions: Using additives that bind to hydrophobic surfaces or increase solvent viscosity.
- Controlling Sample Environment: Precise management of temperature, protein concentration, and buffer composition.
Quantitative Comparison of Anti-Aggregation Agents
The following table summarizes the efficacy and application of common agents used to handle sticky substrates in chaperonin studies.
Table 1: Anti-Aggregation Reagents and Their Properties
| Reagent Class | Example(s) | Typical Working Concentration | Mechanism of Action | Key Considerations for Chaperonin Studies |
|---|---|---|---|---|
| Chemical Chaperones | Arginine, Proline, Betaine (Glycine) | 0.1 - 1.0 M | Preferentially excluded from protein surface, stabilizing native state; Arginine masks hydrophobic and ionic interactions. | High concentrations may inhibit chaperonin-substrate binding. Low concentrations (50-100 mM arginine) often optimal. |
| Non-detergent Sulfobetaines | NDSB-201, NDSB-256 | 0.1 - 1.0 M | Neutral osmolyte that stabilizes proteins without interfering with ionic interactions. | Generally non-interfering with ATPase activity. Useful in refolding buffers. |
| Polyols | Glycerol, Sorbitol | 10-30% (v/v or w/v) | Increases solvent viscosity and stability via preferential exclusion. | High viscosity may slow conformational changes. Common in storage buffers. |
| Detergents | CHAPS, Lauryl Maltose Neopentyl Glycol (LMNG) | 0.1 - 2x CMC | Binds hydrophobic patches, competing with aggregation. | Can denature proteins or disrupt chaperonin function above CMC. Use mild, non-ionic types. |
| Macromolecular Crowders | Ficoll 70, Dextran | 50-100 g/L | Mimics cellular crowding, can enhance folding but may also accelerate aggregation. | Requires empirical optimization. Can affect assay baselines. |
| Reducing Agents | DTT, TCEP | 1-5 mM | Prevents intermolecular disulfide formation. | Essential for cysteine-rich substrates. TCEP is more stable and does not reduce metal ions. |
Detailed Experimental Protocols
Protocol 4.1: Refolding Dilution of a Sticky Substrate for Chaperonin Assays
Objective: Transfer a chemically denatured substrate into a native buffer without aggregation, for use in ATP-dependent folding assays. Materials: Urea-denatured substrate protein (6M Urea, 50 mM Tris-HCl pH 7.5, 5 mM DTT), Refolding Buffer (50 mM Tris-HCl pH 7.5, 50 mM KCl, 10 mM MgCl₂, 1 mM DTT, 0.1 mM EDTA, 10% Glycerol, 100 mM L-Arginine). Procedure:
- Prepare Refolding Buffer and chill on ice.
- Keep the denatured substrate on ice.
- Rapidly dilute the denatured substrate 1:100 into the pre-chilled Refolding Buffer with gentle vortexing.
- Incubate the diluted sample on ice for 15 minutes to allow for partial compaction.
- Centrifuge at 20,000 x g for 10 minutes at 4°C to remove any precipitated aggregates.
- Carefully transfer the supernatant (containing the monodisperse, folding-competent substrate) to a new tube.
- Quantify protein concentration immediately (using A280, correcting for arginine absorbance) and use in chaperonin assays within 1-2 hours.
Protocol 4.2: Monitoring Aggregation Kinetics via Light Scattering
Objective: Quantitatively assess the effectiveness of anti-aggregation strategies. Materials: Purified sticky substrate, Assay Buffer ± additives, Fluorometer or Spectrophotometer. Procedure:
- Prepare substrate protein at 2x desired final concentration (e.g., 2 µM) in a base buffer.
- Prepare 2x solutions of various anti-aggregation additives (e.g., 200 mM Arg, 20% Glycerol) in the same base buffer.
- In a cuvette, mix equal volumes (e.g., 250 µL) of the 2x substrate and 2x additive buffer to initiate the reaction. Final volume 500 µL.
- Immediately place the cuvette in the instrument thermostatted at 25°C.
- Monitor light scattering (90° angle for fluorometer, or absorbance at 360 nm for spectrophotometer) every 30 seconds for 60 minutes.
- Plot scattering intensity vs. time. The initial slope and final plateau are indicators of aggregation rate and extent, respectively.
Visualization of Workflows and Concepts
Diagram Title: Managing Aggregation Pathways for Sticky Substrates
Diagram Title: Experimental Workflow for Substrate Preparation
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Handling Sticky Substrates
| Item | Function/Application | Example Product/Catalog Number |
|---|---|---|
| High-Purity L-Arginine HCl | Primary chemical chaperone to suppress aggregation during dilution/refolding. | MilliporeSigma A5131 |
| TCEP-HCl | Stable, metal-compatible reducing agent to prevent disulfide scrambling. | Thermo Scientific 77720 |
| NDSB-201 | Non-detergent sulfobetaine, a mild anti-aggregation agent. | MilliporeSigma 743027 |
| CHAPS Detergent | Mild zwitterionic detergent for solubilizing hydrophobic patches. | Anatrace C316 |
| Glycerol, Molecular Biology Grade | Stabilizing polyol for storage and assay buffers. | Invitrogen AM9024 |
| Amicon Ultra Centrifugal Filters | Rapid buffer exchange and concentration without sample loss. | Millipore UFC803024 (10 kDa MWCO) |
| Superdex 200 Increase 10/300 GL | Size-exclusion chromatography column for analyzing aggregation state. | Cytiva 28990944 |
| Microcuvettes (Low Binding) | For light scattering/aggregation assays, minimizes surface adhesion. | BrandTech 759150 |
| GroEL/GroES (or TRiC) Purification Kit | Isolated chaperonins for functional assays. | Abcam ab204719 (GroEL) |
Challenges in Reconstituting Functional Complexes from Subunits
The study of macromolecular complex assembly is central to understanding cellular function. Within this field, ATP-dependent chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are essential nanomachines that facilitate the folding and assembly of substrate proteins. A core thesis in chaperonin research posits that these complexes do not merely fold single polypeptide chains but are often indispensable for the correct de novo assembly of oligomeric complexes from their constituent subunits. This whitepaper examines the significant technical challenges inherent in reconstituting functional complexes from purified subunits in vitro, a process that often fails without the physiological context provided by chaperonins. Understanding these challenges is critical for researchers aiming to elucidate assembly mechanisms, model diseases of misassembly, or develop therapeutic interventions targeting complex formation.
Core Challenges inIn VitroReconstitution
Reconstituting a functional complex from its purified subunits faces multiple interdependent hurdles, often explaining the obligate need for chaperonin assistance in vivo.
- Concentration Disparity: Subunit stoichiometry in vivo is tightly regulated. In vitro, achieving and maintaining the correct molar ratios, especially for rare subunits, is difficult. Non-physiological concentrations can drive off-pathway aggregation.
- Kinetic Traps: Folding and assembly are concurrent. An individual subunit may misfold or form unstable intermediates before encountering its partner, becoming assembly-incompetent.
- Co-translational Assembly In Vivo vs. Bulk Mixing In Vitro: In vivo, subunits often assemble co-translationally, minimizing exposure of hydrophobic surfaces. In vitro, fully synthesized, exposed subunits are mixed, increasing aggregation risk.
- Post-Translational Modifications (PTMs): Many complexes require PTMs (phosphorylation, glycosylation) for stability or assembly. Purified subunits may lack necessary PTMs unless expressed in sophisticated systems.
- Chaperonin Dependency: As per the central thesis, many subunits are obligate clients of chaperonins like TRiC. They emerge from the chaperonin cavity in an assembly-competent, "folded-but-not-assembled" state, a condition nearly impossible to mimic in vitro.
Table 1: Quantitative Analysis of Reconstitution Success Rates for Selected Chaperonin-Dependent Complexes
| Complex Name (Organism) | Number of Subunits | Chaperonin Involved | In Vitro Reconstitution Efficiency (Without Chaperonin) | Key Identified Challenge |
|---|---|---|---|---|
| Actin (Human) | Monomer (polymerizes) | TRiC | <5% (prone to aggregation) | Kinetic trapping of folding intermediates; absolute TRiC dependence. |
| α-Tubulin/β-Tubulin Heterodimer (Bovine) | 2 | TRiC | ~1-2% (low yield, incorrect oligomers) | Requires tubulin cofactors (TBCs) after TRiC folding; complex folding landscape. |
| V-ATPase V1 Domain (Yeast) | 8 different subunits | TRiC (for specific subunits) | ~15% (partial activity) | Strict assembly order required; some subunits are TRiC obligate clients. |
| GroEL/GroES (E. coli) | 14 + 7 (HSP60/HSP10) | Self-assembly (post-translational) | ~75% (high efficiency) | Assembly is chaperonin-independent but requires Mg-ATP and K+ ions. |
Detailed Experimental Protocol: Reconstitution of TRiC-Dependent Tubulin Heterodimer
This protocol highlights the multi-step, chaperonin-dependent process required for successful assembly of a fundamental complex.
Objective: To reconstitute functional α/β-tubulin heterodimers from denatured, purified recombinant subunits using the chaperonin TRiC and cofactors.
Materials:
- Denatured recombinant human α-tubulin and β-tubulin.
- Purified human TRiC complex (CCT).
- Purified tubulin cofactors (TBCs: TBCA, TBCB, TBCC, TBCE, etc.).
- Reconstitution Buffer: 50 mM HEPES-KOH (pH 7.4), 50 mM KCl, 5 mM MgCl₂, 1 mM DTT.
- ATP-Regeneration System: 2 mM ATP, 10 mM Creatine Phosphate, 50 µg/mL Creatine Kinase.
- Pre-chilled 96-well plates or thin-wall PCR tubes.
- Thermostated spectrophotometer/fluorometer.
Procedure:
- Chaperonin-Mediated Folding:
- Prepare separate reactions for α and β-tubulin. In a 50 µL volume in Reconstitution Buffer, mix 1 µM denatured tubulin subunit with 0.2 µM TRiC.
- Initiate folding by adding the ATP-regeneration system.
- Incubate at 30°C for 90 minutes to allow complete ATP-driven folding cycles within the TRiC cavity.
Cofactor-Dependent Dimerization:
- Combine the two completed folding reactions.
- Add the necessary tubulin cofactors at stoichiometric ratios (typically 1:1 molar ratio of TBCs to tubulin).
- Continue incubation at 30°C for an additional 60 minutes. This allows TBCs to capture folded monomers and facilitate heterodimer formation.
Release and Purification:
- The heterodimer is released from the TBC complex upon addition of GTP. Add GTP to a final concentration of 1 mM and incubate for 15 min.
- Remove TRiC, TBCs, and unassembled material by size-exclusion chromatography (SEC) or native PAGE.
- Collect the peak corresponding to the ~100 kDa heterodimer.
Functional Validation:
- Polymerization Assay: Monitor turbidity at 350 nm upon warming the heterodimer to 37°C in the presence of GTP and a catalytic amount of pre-formed microtubule seeds.
- GTPase Activity: Measure phosphate release using a malachite green assay.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Complex Reconstitution Studies
| Reagent / Material | Function in Reconstitution Experiments |
|---|---|
| TRiC/CCT (Purified Complex) | Essential chaperonin for folding obligate client subunits like actin/tubulin. Provides the in vivo-like folding environment. |
| ATP-Regeneration System | Maintains constant, high ATP levels during long chaperonin reactions, crucial for multiple folding cycles. |
| Tubulin Cofactor (TBC) Kit | Set of recombinant proteins (TBCA-E) required for the final assembly and release of functional α/β-tubulin heterodimers post-TRiC folding. |
| Site-Specific Fluorophores (e.g., Maleimide-Dyes) | For labeling specific cysteine mutants on subunits to monitor conformational changes or subunit association via FRET. |
| Native Gel Electrophoresis System | Non-denaturing polyacrylamide gels to separate and visualize successfully assembled complexes from sub-complexes or aggregates. |
| Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS) | Determines the absolute molecular weight and oligomeric state of the reconstituted complex in solution, confirming correct assembly. |
| Surface Plasmon Resonance (SPR) Chips | Functionalized with one subunit to measure real-time kinetic parameters (ka, kd) of binding to its partner subunit. |
Visualization of Pathways and Workflows
Diagram 1: Chaperonin-Guided Pathway vs. Aggregation Risk (82 chars)
Diagram 2: Experimental Workflow for Chaperonin-Assisted Reconstitution (88 chars)
Strategies for Differentiating between Assisted Folding and Mere Holdase Activity
Within the broader thesis on ATP-dependent chaperonin mechanisms, a critical and often nuanced challenge is distinguishing genuine, iterative folding promotion from passive, aggregation-preventing holdase activity. This guide provides a technical framework for researchers to design definitive experiments, leveraging kinetic, structural, and single-molecule approaches to dissect these functionally distinct chaperone modes.
Chaperones prevent protein misfolding and aggregation. Holdases bind non-native substrates, suppressing aggregation in an ATP-independent manner. Foldases, particularly ATP-dependent chaperonins like GroEL/ES or TRiC, actively promote conformational rearrangement towards the native state. Misattribution of function can skew mechanistic models and drug discovery efforts targeting proteostasis.
Quantitative Distinguishing Features: Kinetic and Thermodynamic Hallmarks
The core differentiators lie in the effect on the folding trajectory. The following table summarizes key measurable parameters.
Table 1: Comparative Features of Holdase vs. Assisted Folding Activity
| Parameter | Mere Holdase Activity | Assisted Folding Activity | Primary Assay |
|---|---|---|---|
| ATP Dependence | None | Strictly required | Coupled enzymatic ATPase assay. |
| Folding Rate Constant (k_fold) | Unchanged or slightly decreased. | Significantly increased. | Stopped-flow fluorescence/light scattering. |
| Final Native Yield | Increased (prevents aggregation loss). | Increased (prevents loss + enhances folding). | Size-exclusion chromatography, activity assays. |
| Single-Turnover Efficiency | Low; release often yields unproductive substrate. | High; release yields native or folding-committed substrate. | Single-turnover experiment with trap. |
| Effect on Aggregation Kinetics | Suppresses aggregation onset. | Suppresses aggregation and speeds native formation. | Light scattering at 320-360 nm. |
Core Experimental Protocols
Single-Turnover Folding with an Irreversible Trap
Objective: Determine if a chaperone-substrate complex, upon ATP addition, releases a product committed to native folding or an unstable intermediate.
Protocol:
- Complex Formation: Incubate chaperone (e.g., 1 µM GroEL tetradecamer) with chemically denatured, fluorescently labeled substrate (e.g., 2 µM MDH or rhodanese) in refolding buffer (e.g., 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 10 mM MgCl₂) for 5-10 min.
- Trap Introduction: Add a large molar excess (e.g., 20-fold) of an unlabeled, aggregation-prone "trap" protein (e.g., citrate synthase). This trap will immediately bind any free chaperone released during the reaction, preventing rebinding to the labeled substrate.
- Reaction Initiation: Rapidly add ATP (2-5 mM final) and an ATP-regenerating system (e.g., 20 mM creatine phosphate, 50 µg/mL creatine kinase).
- Monitoring: Track labeled substrate fluorescence (for conformation) and/or enzymatic activity recovery over time.
- Interpretation: A holdase will release the labeled substrate upon ATP addition, but it will be captured by free trap protein and likely aggregate or fold slowly. An active foldase will show a rapid, trap-resistant increase in native signal, indicating productive folding completion before chaperone recycling.
Stopped-Flow Kinetics of Aggregation Suppression vs. Folding Promotion
Objective: Decouple the kinetics of aggregation prevention from the acceleration of native state formation.
Protocol:
- Sample Loading: Load two syringes of a stopped-flow apparatus:
- Syringe A: Denatured substrate (e.g., 5 µM luciferase) in refolding buffer +/- chaperone.
- Syringe B: Refolding buffer containing ATP (or non-hydrolysable analog) and ATP-regenerating system.
- Dual-Channel Detection:
- Channel 1: Light scattering at 360 nm (aggregation signal).
- Channel 2: Tryptophan fluorescence or FRET signal (conformational change).
- Rapid Mixing: Initiate refolding by 1:1 mixing at 25°C.
- Data Analysis: Compare traces. A pure holdase will show suppressed light scattering but no change in folding fluorescence kinetics versus spontaneous refolding. An active foldase will show both suppressed scattering and a decreased half-time (t₁/₂) for the fluorescence change.
Visualization of Conceptual and Experimental Frameworks
Title: Decision Tree for Holdase vs. Foldase Outcomes
Title: Stopped-Flow Assay Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Differentiation Studies
| Reagent / Material | Function / Rationale | Example Product / Specification |
|---|---|---|
| ATP-regenerating System | Maintains constant [ATP] during long assays; critical for multi-turnover folding. | Creatine phosphate & creatine kinase. Pyruvate kinase & phosphoenolpyruvate. |
| Non-hydrolysable ATP Analog (e.g., ATPγS, AMP-PNP) | Distinguishes ATP binding effects from hydrolysis/energy transduction. | Specific for testing commitment steps. |
| Bulk Aggregation Trap Protein | Acts as an irreversible sink for free chaperone in single-turnover experiments. | Citrate synthase, malate dehydrogenase (high chaperone affinity). |
| Model Substrate Proteins | Well-characterized, aggregation-prone proteins for benchmarking. | Rhodanese, Firefly Luciferase, Malate Dehydrogenase (MDH). |
| Fluorescent Dyes for Labeling | Site-specific (cysteine-reactive) or non-specific labeling for FRET/conformational tracking. | Alexa Fluor 488/555 maleimide, ANS (hydrophobicity probe). |
| Chaperone Mutants (e.g., ATPase-deficient) | Controls for chaperone mechanochemical activity. | GroEL D398A, Hsp70 K71M. |
| Size-Exclusion Chromatography (SEC) Column | Resolves native monomers from aggregates/chaperone complexes. | Superdex 200 Increase, analytical grade. |
| Stopped-Flow Apparatus | Measures rapid kinetics of folding/aggregation (millisecond resolution). | Equipped with fluorescence and light scattering detection. |
Advanced Correlative Approaches
- Single-Molecule FRET (smFRET): Directly visualizes conformational dynamics of a substrate protein while bound to and released from a chaperone, providing unambiguous proof of foldase-induced remodeling.
- Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS): Maps regions of substrate stabilized or destabilized by chaperone binding, identifying if the interaction preserves non-native structure (holdase) or induces protective compaction (foldase precursor).
- Cryo-EM: Visualizes distinct conformational states of chaperone-substrate complexes in the presence of ATP analogs versus ADP, revealing mechanical actions indicative of active folding.
Definitively differentiating assisted folding from holdase activity requires a multi-parametric approach, combining stringent single-turnover kinetics, correlated aggregation/folding assays, and advanced structural biology. Integrating these strategies is essential for accurately characterizing chaperonin mechanisms and for validating targets in drug development aimed at modulating proteostasis networks.
Best Practices for Data Reproducibility and Validating Folding Efficiency
Within the study of ATP-dependent chaperonin mechanisms, data reproducibility and accurate measurement of protein folding efficiency are paramount. Chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, utilize ATP hydrolysis to drive conformational changes that facilitate native protein folding. This whitepaper outlines rigorous practices to ensure reproducible, validated results in this complex experimental domain.
Section 1: Foundational Principles of Reproducibility
Reproducibility in chaperonin research hinges on meticulous control of biochemical conditions and precise documentation. The inherently dynamic nature of the ATPase cycle and substrate interactions necessitates standardized approaches.
Table 1: Critical Parameters Requiring Strict Documentation for Reproducibility
| Parameter | Typical Range/Value | Impact on Folding Assay | Recommended Documentation |
|---|---|---|---|
| ATP Concentration | 0.1 - 5 mM | Directly drives chaperonin cycle; affects kinetics & yield | Exact stock conc., pH, supplier, aliquot history |
| [Mg2+] (free) | 2 - 10 mM (in excess of ATP) | Essential cofactor for ATP hydrolysis; affects kinetics | Calculated free concentration, considering ATP-Mg chelation |
| Chaperonin Concentration | 0.1 - 1 µM (as oligomer) | Substrate stoichiometry & encapsulation probability | Method of determination (A280, Bradford), oligomeric state verification |
| Substrate Protein Purity & State | >95% purity, urea/GdnHCl denatured | Baseline for % folded calculation; affects aggregation propensity | Denaturant concentration, denaturation time, storage buffer post-denaturation |
| Temperature | 25°C - 37°C | Impacts folding rates, chaperonin ATPase rate | Incubator/block calibrator data, equilibration time |
| Buffer System | e.g., 50 mM Tris-HCl, 50 mM KCl, 10 mM KCl | Ionic strength & pH affect chaperonin stability & kinetics | Exact pH at T, buffer preparation notes, filter sterilization lot |
| Oxidizing/Reducing Environment | 1-10 mM DTT or none | Critical for disulfide-containing substrates | DTT/TCEP fresh preparation, concentration verification |
Section 2: Core Experimental Protocols for Folding Efficiency
Protocol 2.1: Stopped-Flow Kinetic Assay for ATP-Dependent Folding
Objective: Measure the kinetics of substrate folding triggered by ATP/Mg2+ addition to the chaperonin-substrate complex. Methodology:
- Prepare Solutions:
- Syringe A: 2 µM chaperonin (GroEL/TRiC) complexed with 2 µM denatured substrate protein in assay buffer (50 mM Tris-HCl, 100 mM KCl, pH 7.4) with 1 mM DTT. Pre-incubate for 5 minutes.
- Syringe B: Assay buffer containing 5 mM ATP, 10 mM MgCl2, and any co-chaperonin (e.g., GroES at 4 µM).
- Data Acquisition: Load syringes into stopped-flow apparatus thermostatted at 25°C. Rapidly mix equal volumes (typical dead time ~1 ms). Monitor signal change.
- Signal Monitoring:
- For fluorescent substrates: Use intrinsic tryptophan fluorescence (excitation 295 nm, emission 340 nm) or extrinsic label (e.g., ANS).
- For light scattering: Monitor at 320 nm to track aggregation.
- Analysis: Fit resulting kinetic traces to multi-exponential functions. Report amplitudes and rate constants (k_obs). Normalize amplitude to 0% (fully denatured) and 100% (natively refolded control) baselines.
Protocol 2.2: Native Gel Electrophoresis for Folding Yield Quantification
Objective: Quantitatively separate native substrate from misfolded/aggregated species post-chaperonin reaction. Methodology:
- Folding Reaction: Conduct folding assay in a tube (e.g., 1 µM chaperonin, 1 µM substrate, 2 mM ATP, 5 mM MgCl2, buffer). Terminate at defined timepoints (e.g., 0, 5, 30, 60 min) by adding 5 mM EDTA.
- Sample Preparation: Mix terminated reaction 1:1 with native gel loading buffer (50% glycerol, 0.01% bromophenol blue, no SDS or β-mercaptoethanol).
- Electrophoresis: Load on pre-cast 4-16% polyacrylamide gradient native gel. Run in Tris-Glycine buffer (pH 8.3) at 100V for 90 min at 4°C to maintain complexes.
- Detection & Quantification: Stain with Coomassie or Sypro Ruby. Image with a calibrated digital imager. Quantify band intensity for native substrate using ImageJ or similar. Calculate folding yield as (Native band intensity / Total substrate band intensity) x 100%.
Protocol 2.3: ATPase Activity Coupled Assay
Objective: Correlate chaperonin-facilitated folding with ATP hydrolysis activity. Methodology:
- Setup: In a 96-well plate, mix chaperonin (0.2 µM oligomer) with/without substrate protein (0-4 µM) in assay buffer containing 2 mM phosphoenolpyruvate (PEP), 0.2 mM NADH, 10 U/ml pyruvate kinase, and 10 U/ml lactate dehydrogenase.
- Initiation: Start reaction by adding MgATP to a final concentration of 1 mM.
- Monitoring: Follow NADH absorbance at 340 nm every 30s for 30 min at 30°C using a plate reader.
- Calculation: ATP hydrolysis rate is proportional to ΔA340/min. Calculate specific activity (nmol ATP/min/nmol chaperonin). Substrate presence often stimulates ATPase.
Table 2: Summary of Key Assay Outputs for Folding Efficiency
| Assay Type | Primary Readout | Quantifiable Metric | Typical Time Scale | Information Gained |
|---|---|---|---|---|
| Stopped-Flow Kinetics | Fluorescence/Light Scattering | Rate constants (k_fold), Amplitudes | Milliseconds to Minutes | Burst phases, kinetic intermediates, aggregation onset |
| Native Gel | Band Intensity | % Native Yield | Minutes to Hours | Final folding yield, stability of native product, complex disassembly |
| Coupled ATPase | A340 (NADH oxidation) | ATP hydrolyzed / min / chaperonin | Minutes to Hours | Energetic cost of folding, substrate-induced stimulation |
Section 3: Visualizing the Chaperonin Cycle and Experimental Workflow
Title: ATP-Dependent Chaperonin Folding Cycle
Title: Workflow for Validating Chaperonin Folding Efficiency
Section 4: The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Chaperonin Folding Assays
| Reagent / Material | Supplier Examples (for reference) | Key Function & Rationale | Critical Quality Control |
|---|---|---|---|
| Ultra-Pure ATP (Na+/Mg2+ salt) | Sigma-Aldrich, Roche | Primary energy source for chaperonin cycle; impurities inhibit hydrolysis. | HPLC-purified; verify concentration by A259 (ε=15,400 M⁻¹cm⁻¹). Aliquot, store at -80°C. |
| Chaperonin Proteins (GroEL/ES, TRiC) | Home-purified (recommended), commercial (e.g., AdipoGen) | Core machinery. Must be >95% pure, oligomerically homogeneous (14-mer/16-mer). | Verify oligomeric state via native PAGE or SEC-MALS. Test basal ATPase activity. |
| Model Substrate Proteins | e.g., Rhodanese, MDH, Firefly Luciferase | Well-characterized, hard-to-fold proteins for benchmarking folding efficiency. | Ensure complete, controlled denaturation (e.g., in 6M GdnHCl). Verify lack of spontaneous refolding. |
| Coupling Enzymes (PK/LDH) | Roche, Sigma | For coupled ATPase assay; convert ADP to ATP, oxidizing NADH proportionally. | Check for ammonium sulfate precipitation; dialyze before use to avoid carryover. |
| Fluorescent Dyes (ANS, Bis-ANS) | Thermo Fisher, Molecular Probes | Hydrophobic dyes reporting on exposed hydrophobic surfaces (misfolded states). | Titrate to determine optimal concentration; avoid inner filter effect. |
| Size-Exclusion Chromatography (SEC) Columns | Cytiva (Superose 6 Increase), Tosoh Bioscience | Analytical separation of chaperonin complexes, folded vs. unfolded substrates. | Calibrate with known standards; use same column lot for comparative studies. |
| Stable Denaturants (Urea/GdnHCl) | Millipore, Sigma | For reproducible, complete substrate unfolding. Critical for baseline definition. | Use ultrapure grade; determine concentration by refractometry. Filter to remove cyanate (urea). |
| Reducing Agents (TCEP, DTT) | GoldBio, Thermo Fisher | Maintain reduced state for cysteines; prevent non-native disulfide formation. | TCEP is more stable than DTT. Prepare fresh stocks, pH-adjusted. Confirm concentration. |
Section 4: Validation and Orthogonal Approaches
Robust validation requires converging evidence from multiple techniques. Correlate folding kinetics (stopped-flow) with final yield (native gel). Cross-validate functional activity recovery (e.g., enzymatic activity of folded substrate) with structural assessments (e.g., CD spectroscopy, limited proteolysis). Always include positive (spontaneous folding if any) and negative (no ATP, chaperonin mutant) controls. Data from ATPase assays should align temporally with kinetic phases of folding. Document all metadata, including raw data files, analysis scripts (e.g., Python, R), and instrument calibration logs, adhering to FAIR (Findable, Accessible, Interoperable, Reusable) data principles to ensure full reproducibility and robust validation of folding efficiency within the ATP-dependent chaperonin mechanism thesis.
Validation and Context: How Chaperonins Compare in the Cellular Chaperone Network
Within the broader thesis on ATP-dependent mechanisms of chaperonins, the GroEL/ES system remains the quintessential model for understanding how ring-shaped molecular machines utilize ATP hydrolysis to drive conformational changes essential for protein folding. This whitepaper consolidates the key experimental evidence validating the ATP-driven cyclical model, focusing on quantitative data, definitive protocols, and the tools that enabled these discoveries.
Table 1: Key Quantitative Parameters of the ATP-Driven GroEL/ES Cycle
| Parameter | Experimental Value | Measurement Technique | Functional Implication |
|---|---|---|---|
| ATP Hydrolysis Rate (per ring) | ~85 s⁻¹ at 25°C | Coupled enzymatic assay (NADH oxidation) | Defines cycle tempo; positive cooperativity within ring. |
| ATP Binding Affinity (Kd) | ~10-30 µM (T-state); <1 µM (R-state) | Tryptophan fluorescence quenching, ITC | Allosteric switch from low to high affinity upon ATP binding. |
| Conformational Change Timescale (T to R) | ~5-20 ms | Stopped-flow FRET (Cys-labeled GroEL) | Rapid rigid-body domain movements enabling substrate release. |
| Substrate Protein Encapsulation Time | ~100-200 ms (post ATP binding) | Single-molecule FRET, Cryo-EM time-resolved | Coordinated with GroES binding; creates folding chamber. |
| Folding Chamber Residence Time | ~10-15 seconds | Radiolabeled substrate release assays | Defines the timeframe for passive, isolated folding. |
| Energy Cost per Folded Polypeptide | ~100-150 ATP molecules | Calorimetry & substrate yield quantification | Highlights stoichiometric vs. catalytic chaperonin action. |
Table 2: Critical Mutagenesis & Inhibitor Studies
| Intervention | Target | Observed Phenotype | Evidence for ATP-Drive |
|---|---|---|---|
| GroEL(D398A) Mutant | ATP hydrolysis (Walker B motif) | Binds ATP, no hydrolysis; complex stalls with GroES & substrate trapped. | Proof that hydrolysis is essential for cycle progression and trans ring activation. |
| GroEL(R13G/A26V) Double Mutant | Inter-ring negative allostery | Eliminates trans ring communication; rings hydrolyze ATP independently. | Validates the alternating cycle model; negative cooperativity is a core design feature. |
| AMP-PNP (Non-hydrolyzable Analog) | ATP binding pocket | Induces GroES binding & encapsulation but blocks discharge. | Separates ATP binding effects from hydrolysis; demonstrates ATP binding is sufficient for major conformational change. |
| AlFx (Transition State Analog) | Post-hydrolysis state (ADP·Pi) | Traps a hydrolysis-committed state, stabilizing the cis folding complex. | Pinpoints the role of hydrolysis products in triggering subsequent steps. |
Key Experimental Protocols
Stopped-Flow FRET for Monitoring Ring-Switching Kinetics
Objective: To measure the rate of allosteric signal transmission between the two GroEL rings upon ATP binding. Protocol:
- Labeling: Introduce unique surface-exposed cysteine residues via site-directed mutagenesis in each ring (e.g., S90C in one ring, S516C in the other). Label separately with donor (e.g., Alexa Fluor 488 maleimide) and acceptor (e.g., Alexa Fluor 594 maleimide) fluorophores.
- Separation: Purify single-labeled heptameric rings by ion-exchange chromatography and reconstitute asymmetric "hetero-double-ring" GroEL by controlled annealing.
- Experiment: Load the labeled GroEL into one syringe of a stopped-flow apparatus. Load ATP (with or without GroES/substrate) into the second syringe.
- Measurement: Rapidly mix (dead time ~1 ms) and monitor acceptor emission upon donor excitation. The FRET signal reports on the relative orientation/distance between the rings.
- Analysis: Fit the resulting fluorescence transient to exponential functions. The observed rate constant quantifies the trans-to-cis ring switching triggered by ATP.
Single-Molecule Substrate Encapsulation Assay
Objective: To directly visualize the timing and efficiency of substrate protein encapsulation upon ATP/GroES binding. Protocol:
- Substrate Labeling: Engineer a cysteine into a well-folded substrate protein (e.g., rhodanese) and label with a bright fluorophore (e.g., Cy5).
- Surface Immobilization: Biotinylate GroEL via a introduced surface lysine. Attach to a PEG-passivated, streptavidin-coated quartz microscope slide.
- Imaging: Use total internal reflection fluorescence (TIRF) microscopy. Introduce flowing solution containing labeled substrate, GroES, and ATP (with oxygen scavenging system for photostability).
- Data Acquisition: Track the appearance and lifetime of single Cy5 spots (substrate) colocalized with immobilized GroEL. A sudden increase in fluorescence intensity followed by a stable period indicates encapsulation (protected from quenching).
- Analysis: Generate histograms of encapsulation lag times and residence durations, directly providing the quantitative parameters in Table 1.
Visualization of Mechanisms and Workflows
Title: ATP-Driven GroEL/ES Functional Cycle
Title: FRET Experiment for Ring Switching Kinetics
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for ATP-Driven Mechanism Validation
| Reagent/Category | Specific Example(s) | Function & Rationale |
|---|---|---|
| Non-hydrolyzable ATP Analogs | AMP-PNP, ATPγS | To dissect conformational changes induced by ATP binding from those requiring hydrolysis. Essential for trapping intermediate states. |
| Hydrolysis-Deficient Mutants | GroEL(D398A), GroEL(E409A) | Genetically "clamp" the chaperonin cycle at specific points to study structural and functional intermediates. |
| Allosteric Mutants | GroEL(R13G/A26V), GroEL(Y203A) | Disrupt inter-ring communication to prove negative cooperativity is essential for the alternating cycle. |
| Fluorescent Nucleotides | Mant-ATP (2'/3'-O-(N-Methylanthraniloyl)), TNP-ATP | Directly monitor nucleotide binding kinetics and affinity via fluorescence enhancement/quenching. |
| Site-Specific Labeling Kits | Maleimide derivatives of Alexa Fluor dyes, HaloTag ligands | For introducing FRET pairs or fluorescent probes at precise locations to track domain movements. |
| Rapid Kinetics Instruments | Stopped-Flow Spectrofluorometer, Quench-Flow Apparatus | To capture fast (ms-timescale) conformational changes and ATP hydrolysis events. |
| Single-Molecule Imaging Buffers | Glucose Oxidase/Catalase "GLOX" system, Trolox | Oxygen scavenging and triplet-state quenching systems essential for prolonged, stable single-fluorophore observation. |
| Trapped Intermediate Stabilizers | Aluminum Fluoride (AlFx), Beryllium Fluoride (BeFx) | Mimic the γ-phosphate transition state, stabilizing post-hydrolysis complexes (e.g., ADP·Pi state) for structural analysis. |
| Native Substrate Proteins | Rhodanese, MDH (Mitochondrial Malate Dehydrogenase) | Well-characterized, stringent chaperonin substrates that reliably unfold and require GroEL/ES for efficient in vitro refolding. |
This whitepaper provides a quantitative and mechanistic comparison of ATP consumption across three major chaperone systems: the barrel-shaped chaperonins (e.g., GroEL/GroES, TRiC/CCT), Hsp70/DnaK, and Hsp90. A core thesis in chaperonin research posits that these complexes are high-energy "power users" of ATP, employing concerted, allosteric hydrolysis cycles to drive large-scale conformational changes essential for substrate folding. This analysis contrasts that mechanism with the more iterative, stochastic ATPase cycles of Hsp70 and the structurally restrictive, ATP-triggered clamping of Hsp90, evaluating the energetic cost-efficiency of each system in cellular proteostasis.
Quantitative Comparison of ATPase Parameters
Table 1: Comparative ATP Hydrolysis Metrics of Major Chaperone Systems
| Parameter | Chaperonins (GroEL/GroES) | Hsp70 (DnaK System) | Hsp90 |
|---|---|---|---|
| ATP Molecules per Cycle | 7 (per ring); 14 (full complex) | 1 (per monomer) | 2 (per dimer) |
| Hydrolysis Rate (min⁻¹) | ~10-15 (per ring) | ~0.02-0.2 (basal); ~1-5 (stimulated) | ~1-2 (basal); ~10-30 (stimulated) |
| Primary Regulator | Co-chaperonin (GroES) encapsulation | Nucleotide Exchange Factors (GrpE), J-domain proteins (DnaJ) | Co-chaperones (Aha1, p23), Client binding |
| Energetic Cost per Client | High (14 ATP/folding cycle) | Variable (Low-High, iterative cycles) | Moderate-High (with co-chaperone cascade) |
| Conformational Scope | Massive, concerted domain rotations and apical lid elevation | Localized substrate-binding domain (SBD) docking to NBD | Dramatic dimer twister-and-clamp motions |
Data compiled from recent studies (2021-2024). Rates are approximate and condition-dependent.
Detailed Experimental Protocols for ATPase Analysis
Protocol 1: Coupled Enzymatic ATPase Assay (Standard for All Systems)
- Objective: Measure steady-state ATP hydrolysis kinetics.
- Reagents: ATP-regeneration system (phosphoenolpyruvate, pyruvate kinase, lactate dehydrogenase), NADH, target chaperone, relevant co-chaperones/substrate.
- Procedure:
- Prepare assay buffer (e.g., 50 mM HEPES-KOH, pH 7.5, 100 mM KCl, 10 mM MgCl₂).
- Mix chaperone (0.1-1 µM) with regeneration system (1 mM phosphoenolpyruvate, 20 U/ml pyruvate kinase, 20 U/ml lactate dehydrogenase), 0.2 mM NADH.
- Initiate reaction by adding ATP (typically 1-5 mM).
- Monitor NADH oxidation at 340 nm (ε = 6220 M⁻¹cm⁻¹) continuously for 10-30 min using a spectrophotometer.
- Calculate hydrolysis rate from the linear slope, correcting for background.
Protocol 2: Single-Turnover ATP Hydrolysis with Rapid Quenched-Flow
- Objective: Measure the intrinsic hydrolysis rate of a pre-formed chaperone-ATP complex.
- Procedure:
- Pre-incubate chaperone (5-10 µM) with trace [γ-³²P]ATP in one syringe.
- Rapidly mix with a large excess of unlabeled ATP (chase) and quenching solution (5% formic acid) in a quenched-flow apparatus (mixing time < 100 ms).
- Analyze time points (ms to s) by thin-layer chromatography to quantify hydrolyzed ³²P~Pi.
- Fit data to a single exponential to determine the first-order hydrolysis rate constant (k_hyd).
Protocol 3: Biolayer Interferometry (BLI) for Real-Time ATPase-Conformation Linkage
- Objective: Correlate ATP binding/hydrolysis with conformational changes.
- Procedure:
- Immobilize biotinylated chaperone onto streptavidin BLI biosensors.
- Dip sensors into solutions containing ATP or analogs (ATPγS, ADP, AMP-PNP).
- Monitor wavelength shift in real-time, which reflects mass/structural change.
- Perform parallel assays with hydrolysis-deficient mutants to decouple binding from hydrolysis-driven steps.
Signaling Pathways and Workflow Visualizations
Diagram 1: ATP-Driven Chaperone Functional Cycles (76 chars)
Diagram 2: Experimental Workflow for ATP Usage Comparison (79 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Chaperone ATPase Studies
| Reagent / Material | Function & Rationale |
|---|---|
| Recombinant Chaperone Proteins | Purified, active GroEL/ES, Hsp70, Hsp90 systems. Basis for in vitro biochemistry. |
| Nucleotide Analogs (ATPγS, AMP-PNP, ADP-BeFx) | Hydrolysis-resistant or transition-state mimics to trap specific conformational states. |
| Coupled Enzyme System (PEP/PK/LDH) | Enables continuous, spectrophotometric monitoring of ATP turnover in real-time. |
| [γ-³²P]ATP or [α-³²P]ATP | Radiolabeled ATP for high-sensitivity, single-turnover or phosphate release assays. |
| J-domain Protein (DnaJ) & NEF (GrpE) | Essential cofactors for stimulating and regulating Hsp70 ATPase cycle. |
| Hsp90 Co-chaperones (Aha1, p23) | Potent stimulators and modulators of Hsp90 ATPase cycle and client maturation. |
| Model Denatured Substrate (e.g., Rhodanese, Luciferase) | Standardized client protein to measure chaperone-dependent refolding and ATP coupling. |
| Size-Exclusion Chromatography (SEC) Columns | Critical for separating nucleotide-bound states and chaperone-cofactor complexes. |
Cellular proteostasis is governed by a network of molecular chaperones, a significant subset of which are ATP-dependent. The broader thesis of contemporary chaperonin research posits that the ATPase cycle drives conformational changes essential for substrate protein (client) binding, encapsulation, folding, and release. Within this network, a fundamental question arises: how do different chaperone families and isoforms, often with overlapping in vitro substrate spectra, achieve specificity and partition substrates in vivo? This whitepaper examines the mechanisms of functional overlap and specificity among major ATP-dependent chaperone systems—including Hsp70, Hsp90, and the chaperonins (GroEL/GroES, TRiC)—focusing on how substrate partitioning is regulated to ensure efficient proteostasis and prevent aggregation.
Core Chaperone Systems: Roles and ATP-Driven Cycles
Hsp70 (DnaK): Binds short hydrophobic peptide segments in an ATP-controlled manner. ATP-bound state has low affinity and high exchange rates; ATP hydrolysis to ADP increases affinity, stabilizing client binding. Nucleotide exchange factors (NEFs) catalyze ADP release, resetting the cycle. Co-chaperones like J-domain proteins (JDPs) target Hsp70 to specific clients and stimulate ATPase activity.
Hsp90: A dimeric chaperone that undergoes a large ATP-driven conformational cycle from an "open" to a "closed" state. It acts later in the folding process, often on specific, signal-transduction clients (kinases, steroid receptors). Its function is heavily modulated by a cohort of co-chaperones (e.g., Cdc37, Aha1, p23) that dictate client entry, maturation, and release.
Group I Chaperonins (GroEL/GroES): A double-ring complex with a central cavity. ATP binding and hydrolysis in one ring promotes client encapsulation under the co-chaperonin GroES, providing an isolated chamber for folding. The cycle alternates between rings (positive cooperativity within a ring, negative cooperativity between rings).
Group II Chaperonins (TRiC/CCT): A hetero-oligomeric complex with eight different subunits per ring. Does not require a GroES-like cofactor; its lid is built-in. Folds actins, tubulins, and other complex proteins. Subunit specificity is believed to guide client recognition and positioning.
Table 1: Key Characteristics of Major ATP-Dependent Chaperone Systems
| Chaperone System | Primary Clients | ATPase Cycle Role | Key Co-chaperones/Regulators | Typical Cellular Role |
|---|---|---|---|---|
| Hsp70 (DnaK) | Nascent chains, unfolded proteins | Controls substrate binding/release kinetics | J-domain proteins (JDPs), NEFs (GrpE, BAG) | Holdase, early folding, translocation |
| Hsp90 | Kinases, steroid receptors, E3 ligases | Drives conformational "clamp" for client maturation | Cdc37, Aha1, p23, immunophilins | Late-stage activation & stabilization |
| GroEL/GroES | ~10-60 kDa proteins, bacterial substrates | Powers encapsulation in folding cage | GroES (obligate co-chaperonin) | De novo folding, aggregation rescue |
| TRiC/CCT | Actins, tubulins, cell cycle regulators | Coordinates folding in closed chamber | Prefoldin (client delivery), PhLP | Folding of complex, multi-domain proteins |
Mechanisms of Substrate Partitioning: Overlap vs. Specificity
Substrate partitioning is not solely dictated by intrinsic client affinity but is a regulated process involving cellular localization, co-chaperone networks, temporal expression, and the ATP-driven cycle kinetics.
3.1. Co-chaperone-Directed Specificity: Co-chaperones are primary determinants of substrate flux. For example, the JDP family in eukaryotes (DNAJA/B/C) directs Hsp70 to distinct subcellular locales and client pools. In Hsp90, Cdc37 is essential for kinase client recruitment, while Aha1 accelerates the ATPase cycle.
3.2. Kinetic Partitioning via ATP Cycles: The different rates of ATP hydrolysis and conformational changes create "kinetic gates." A rapidly binding but slowly hydrolyzing chaperone (e.g., Hsp70) can hold a substrate, which may then be transferred to a slower-binding but processive chamber (e.g., GroEL) based on relative ATPase cycle timing and affinity.
3.3. Sequential Pathways and Handoffs: Chaperones often act in ordered pathways. A canonical route involves Hsp70 (holding) → Hsp90 (maturation) for signaling proteins. For cytoskeletal proteins, prefoldin delivers clients to TRiC. This handoff minimizes misfolding and aggregation during transfer.
3.4. Cellular Compartmentalization and Stress Response: Partitioning is spatial. Mitochondrial Hsp70 (mtHsp70) and Hsp60/Hsp10 do not overlap with cytosolic systems. During heat shock, upregulated Hsp70 may buffer a broader client range, temporarily overriding basal specificity.
Table 2: Quantitative Parameters Influencing Substrate Partitioning
| Parameter | Hsp70 System | Hsp90 System | GroEL/ES System | TRiC System |
|---|---|---|---|---|
| Approx. Cellular Abundance | 1-5% total protein (stress) | 1-2% cytosolic protein | ~1-3% in E. coli | ~0.5-1% cytosolic protein |
| ATP Turnover Rate (k~cat~, min⁻¹) | 1-10 (regulated by JDPs) | 1-3 (regulated by Aha1/p23) | ~15 per ring (cooperative) | ~2-5 per complex |
| Typical Client Load | Hundreds to thousands | ~200 known clients | ~10-15% of E. coli proteome | ~5-10% of cytosolic proteome |
| Key Affinity Range (K~d~) | 0.1-5 µM (ADP-state) | nM-µM (client dependent) | Low µM (for unfolded state) | nM-µM (specific subunit interactions) |
Experimental Protocols for Studying Chaperone-Substrate Interactions
4.1. Co-Immunoprecipitation (Co-IP) with ATP Analogs:
- Purpose: To capture transient, ATP-cycle-dependent chaperone-client complexes.
- Methodology:
- Lyse cells in mild, non-denaturing buffer (e.g., 40 mM HEPES, 100 mM KCl, 5 mM MgCl₂, 1% Triton X-100) supplemented with protease/phosphatase inhibitors.
- To trap specific states, add non-hydrolyzable ATP analogs (e.g., ATPγS, AMP-PNP) at 5 mM or ADP at 2 mM. Include apyrase (an ATP/ADP hydrolyzing enzyme) as a negative control.
- Pre-clear lysate with protein A/G beads for 30 min at 4°C.
- Incubate lysate with antibody against the chaperone of interest (e.g., anti-Hsp90) conjugated to beads for 2-4 hours at 4°C.
- Wash beads 3-5 times with lysis buffer containing the respective nucleotide.
- Elute bound proteins with Laemmli buffer, analyze by SDS-PAGE and Western blotting for suspected clients.
4.2. Surface Plasmon Resonance (SPR) for Binding Kinetics:
- Purpose: To measure real-time affinity (K~D~), association (k~on~), and dissociation (k~off~) rates under different nucleotide conditions.
- Methodology:
- Immobilize purified chaperone (e.g., Hsp70) on a CMS sensor chip via amine coupling.
- Use HBS-EP buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4) supplemented with 2 mM MgCl₂ and either 1 mM ATP or ADP.
- Inject a series of concentrations of purified client protein or peptide over the chip surface.
- Regenerate the surface between runs with a mild acid (10 mM glycine, pH 2.0) or high salt.
- Fit the resulting sensograms to a 1:1 Langmuir binding model to derive kinetic constants.
4.3. FRET-Based ATPase Cycle Assay:
- Purpose: To visualize how client or co-chaperone binding affects chaperone conformation and ATPase cycle in real time.
- Methodology:
- Engineer a FRET pair (e.g., CFP/YFP) into specific domains of a chaperone (e.g., Hsp90's N- and M-domains).
- Purify the FRET-labeled chaperone.
- In a fluorometer, mix chaperone with ATP and record FRET signal over time.
- Repeat experiment adding purified client protein or co-chaperone (e.g., Aha1).
- A change in FRET efficiency indicates a conformational shift. Correlate with ATP hydrolysis rates measured via coupled enzymatic assays.
Visualizing Chaperone Networks and Pathways
Diagram 1: Substrate partitioning and chaperone handoff pathways.
Diagram 2: Generalized ATPase cycle driving chaperone affinity states.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Chaperone-Substrate Partitioning Research
| Reagent / Material | Supplier Examples | Function & Application |
|---|---|---|
| Non-hydrolyzable ATP Analogs (ATPγS, AMP-PNP) | Sigma-Aldrich, Jena Bioscience | Traps chaperones in ATP-bound conformational state for Co-IP or structural studies. |
| Recombinant Chaperone/Client Proteins | Abcam, Sino Biological, in-house expression | Purified components for in vitro binding, ATPase, and folding assays. |
| Hsp90 Inhibitor (Geldanamycin) | Tocris, MedChemExpress | Specific ATP-competitive inhibitor to probe Hsp90-dependent client maturation in vivo. |
| FRET-Compatible Fluorophore Pairs (CFP/YFP) | Chromotek, Addgene | For engineering biosensors to monitor real-time conformational changes in chaperones. |
| Anti-Tag Antibodies (Anti-FLAG, Anti-His) | GenScript, Thermo Fisher | For immunoprecipitation of tagged chaperones or clients in interaction studies. |
| ATPase/GTPase Assay Kit | Promega, Cytoskeleton | Coupled enzymatic assay to quantitatively measure chaperone ATP hydrolysis rates. |
| Proteasome Inhibitor (MG-132) | Peptide Institute, Selleckchem | Blocks degradation, allowing accumulation of chaperone clients for easier detection. |
| Size-Exclusion Chromatography Columns | Cytiva (Superdex) | To separate and analyze stable chaperone-client complexes from in vitro reconstitutions. |
Chaperonins, specifically the Group I (GroEL/GroES in bacteria, Hsp60/Hsp10 in eukaryotes) and Group II (TRiC/CCT) families, are ATP-dependent molecular machines essential for the de novo folding and refolding of a subset of cellular proteins. Within the broader thesis of ATP-dependent chaperonin research, their dysfunction is critically implicated in disease pathogenesis. In neurodegeneration, failure to facilitate proper folding or disaggregation of key proteins leads to toxic accumulation. In cancer, chaperonins are often co-opted to stabilize oncoproteins and support the proteostatic demands of rapid proliferation and metastasis. This whitepaper details the mechanistic roles, experimental evidence, and research tools central to this field.
Quantitative Data on Chaperonin Dysregulation in Disease
Table 1: Chaperonin Expression and Genetic Alterations in Human Disease
| Disease Context | Chaperonin Complex | Alteration / Expression Level (vs. Control) | Associated Client Proteins | Key Functional Consequence |
|---|---|---|---|---|
| Alzheimer's Disease | Hsp60 (mitochondrial) | ↓ Up to 60% in vulnerable neurons | Mitochondrial complex subunits, tau | Mitochondrial dysfunction, ROS increase |
| Huntington's Disease | TRiC/CCT | Subunit expression altered; CCT5 ↓ 40% | Mutant Huntingtin (mHtt) with polyQ expansion | Reduced mHtt aggregation suppression |
| Parkinson's Disease | Hsp60/Hsp10 | ↑ 2-3 fold in CSF as potential biomarker | α-synuclein (indirect) | Compensatory stress response |
| Multiple Cancers (e.g., Glioma, HCC) | TRiC/CCT | Subunit overexpression (e.g., CCT2 ↑ 5-8 fold) | Cell cycle proteins (e.g., cyclin E, p53), oncogenic kinases | Stabilization of oncoproteins, enhanced proliferation |
| Colorectal Cancer | Hsp60 | ↑ 4-6 fold in tumor tissue | β-catenin, survivin | Inhibition of apoptosis, promotion of invasion |
Table 2: Key ATP-Dependent Functional Parameters of Chaperonins
| Chaperonin Complex | ATP Molecules per Folding Cycle | Cycle Time (approx.) | Key Allosteric Regulation | Disease-Linked Mutant Effect |
|---|---|---|---|---|
| Group I: Hsp60 (GroEL) | 14 ATP (7 per ring) | 10-15 seconds | Positive intra-ring, negative inter-ring cooperativity | Hereditary Spastic Paraplegia (SPG13) mutants show ↓ ATPase & folding |
| Group II: TRiC/CCT | 16 ATP (8 distinct subunits/ring) | 20-30 seconds | Sequential, stochastic ATP hydrolysis | CCT5 mutation in sensory neuropathy disrupts subunit stability |
Detailed Experimental Protocols
Protocol: Assessing Chaperonin-Mediated Suppression of AggregationIn Vitro
Objective: Quantify the ability of TRiC/CCT to suppress the aggregation of amyloidogenic proteins like mutant huntingtin (mHtt).
- Protein Purification: Purify recombinant TRiC complex from bovine testes or an overexpression yeast system. Express and purify mHtt exon1 fragment with expanded polyQ tract (e.g., Q53) tagged with a fluorescent protein (e.g., GFP).
- Aggregation Reaction: Prepare a 100 µL reaction in aggregation buffer (40 mM HEPES, pH 7.4, 150 mM KCl). Use 5 µM mHtt-Q53 alone (negative control) or with TRiC (at molar ratios from 0.5:1 to 2:1 TRiC:mHtt). Include an ATP-regeneration system (2 mM ATP, 10 mM creatine phosphate, 20 µg/mL creatine kinase).
- Kinetic Measurement: Load reaction into a 96-well plate. Monitor aggregation in real-time using a plate reader with fluorescence polarization (FP) (Ex/Em 485/535 nm for GFP) or Thioflavin T (ThT) fluorescence (Ex/Em 440/485 nm) at 37°C with orbital shaking. Take readings every 5 minutes for 12-24 hours.
- Data Analysis: Plot fluorescence vs. time. Calculate the elongation rate and lag time. Compare conditions to determine the concentration-dependent inhibitory effect of TRiC.
Protocol: Co-Immunoprecipitation (Co-IP) to Identify Chaperonin-Cancer Client Interactions
Objective: Identify novel oncogenic clients of the CCT complex in lung cancer cell lines.
- Cell Lysis: Culture relevant cells (e.g., A549). Harvest and lyse in mild lysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 5% glycerol, 1 mM EDTA) supplemented with ATP (1 mM) and protease/phosphatase inhibitors to preserve native interactions.
- Immunoprecipitation: Pre-clear lysate with protein A/G beads. Incubate 1 mg of lysate with 2 µg of anti-CCT2 (or other subunit) antibody or species-matched IgG control overnight at 4°C with gentle rotation.
- Bead Capture: Add protein A/G magnetic beads for 2 hours. Wash beads stringently 4 times with lysis buffer.
- Elution and Analysis: Elute bound proteins with 2X Laemmli buffer. Analyze by Western blot for suspected clients (e.g., cyclin E, KRAS) or by mass spectrometry (LC-MS/MS) for unbiased identification.
Protocol: Assessing the Impact of Chaperonin Inhibition on Cancer Cell Viability
Objective: Determine IC50 of a putative CCT inhibitor (e.g., CT20p peptide) using a viability assay.
- Cell Seeding: Seed 3000 cells/well of a cancer cell line (e.g., HeLa) in a 96-well plate.
- Compound Treatment: After 24 hours, treat with serial dilutions of the inhibitor (e.g., 0.1 µM to 100 µM) in triplicate. Include DMSO-only wells as control.
- Viability Assay: Incubate for 72-96 hours. Add CellTiter-Glo reagent (Promega) to measure ATP content as a proxy for viable cells. Record luminescence.
- Calculation: Normalize luminescence to DMSO control. Plot log(inhibitor) vs. normalized response to calculate IC50 using a four-parameter logistic curve.
Diagrams of Signaling Pathways and Workflows
Title: Chaperonin Failure in Neurodegenerative Protein Aggregation
Title: Oncoprotein Stabilization by CCT in Cancer
Title: Workflow to Identify Disease-Relevant Chaperonin Clients
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Chaperonin-Disease Research
| Item | Function/Application in Research | Example Product/Supplier |
|---|---|---|
| Recombinant Chaperonin Proteins | In vitro folding/aggregation assays; structural studies. | Human TRiC/CCT complex (Sigma-Aldrich, C7683); Recombinant GroEL/GroES (Enzo). |
| Chaperonin-Specific Antibodies | Immunoprecipitation, Western blot, immunohistochemistry to assess expression/localization. | Anti-Hsp60 antibody [clone LK1] (Abcam, ab190828); Anti-CCT2 antibody (Santa Cruz, sc-373966). |
| ATP-Regeneration System | Maintains ATP levels in in vitro and cell-based assays to study functional chaperonin cycles. | ATP, Creatine Phosphate, Creatine Kinase kit (Roche, 10127531001). |
| Aggregation-Sensing Dyes | Real-time quantification of amyloid or aggregate formation in inhibition assays. | Thioflavin T (Sigma, T3516); ProteoStat Aggregation Assay (Enzo, ENZ-51023). |
| Chaperonin Inhibitors/Modulators | Tool compounds to probe chaperonin function in disease models. | CCT inhibitor HSF1A; Peptide-based inhibitor CT20p (research-grade, Tocris). |
| Proteasome Inhibitor (Control) | Used to distinguish between chaperonin-mediated folding and proteasomal degradation pathways. | MG-132 (Sigma, C2211). |
| Live-Cell ATP Assay | Measure cell viability as a functional readout of chaperonin inhibition in cancer cells. | CellTiter-Glo Luminescent Viability Assay (Promega, G7570). |
| Proximity Ligation Assay (PLA) Kits | Detect in situ protein-protein interactions between chaperonins and client proteins in fixed cells/tissues. | Duolink PLA (Sigma, DUO92101). |
This whitepaper provides a technical framework for assessing the druggability of chaperonin targets, specifically their ATPase site and allosteric pockets. This investigation is situated within the broader thesis of elucidating the ATP-dependent mechanisms of chaperonins—large, cylindrical complexes that facilitate protein folding in an ATP-hydrolysis-dependent manner. Dysregulation of chaperonin function is implicated in cancer, neurodegenerative diseases, and protein aggregation disorders, making them compelling but challenging therapeutic targets. Effective pharmacological intervention requires a dual-strategy assessment: targeting the conserved, active ATPase site for competitive inhibition and identifying functionally critical allosteric pockets for more selective modulation.
Chaperonins, such as Group I (GroEL in bacteria, Hsp60 in mitochondria) and Group II (TRiC/CCT in eukaryotes), operate through a concerted cycle of ATP binding, hydrolysis, and cooperative conformational changes. The ATPase site is located at the equatorial domain of each subunit. Key features relevant to druggability include:
- High Conservation: The catalytic site is structurally similar across families, raising challenges for selectivity, especially between human cytosolic (TRiC) and mitochondrial (Hsp60) complexes.
- Dynamic Nature: The site undergoes significant conformational shifts between nucleotide states (apo, ATP-bound, ADP-bound), which can be exploited for state-selective inhibition.
- Key Residues: Typically involve Walker A (P-loop) and Walker B motifs, a catalytic base (often aspartate), and sensor residues coordinating the γ-phosphate.
Table 1: Quantitative Metrics for Representative Chaperonin ATPase Sites
| Chaperonin | Group | ATP Kd (μM) | kcat (s-1) | Key Coordinating Residues | Estimated Pocket Volume (ų) |
|---|---|---|---|---|---|
| GroEL (E. coli) | I | 10-30 | ~0.1-0.3 | Asp52, Lys51, Thr30, Glu309 | ~450 |
| TRiC/CCT (Human) | II | 5-15 | ~0.05-0.1 | Asp/Asp/Glu (varies by subunit), Lys, Ser | ~400-550* |
| Hsp60 (Human Mitochondrial) | I | 15-40 | ~0.1-0.2 | Asp83, Lys51, Thr31, Glu322 | ~470 |
*Volume varies among the eight paralogous subunits.
Experimental Protocols for Druggability Assessment
Protocol 3.1: In Silico Druggability Screening of the ATPase Site
Objective: To computationally predict and rank small molecules for binding to the ATPase pocket across different conformational states. Methodology:
- Structure Preparation: Retrieve PDB files (e.g., 1AON for apo-GroEL, 4PKO for ATP-bound TRiC). Process with molecular modeling software (e.g., Schrödinger Maestro, UCSF Chimera): add hydrogens, assign bond orders, optimize H-bond networks.
- Pocket Definition: Define the binding site using the coordinates of bound ATP/ADP, expanded by a 5-8 Å radius.
- Molecular Docking: Perform high-throughput virtual screening of libraries (e.g., ZINC, Enamine) using Glide SP or AutoDock Vina. Dock against multiple conformational snapshots from molecular dynamics (MD) simulations.
- Scoring & Ranking: Compounds are ranked by docking score (kcal/mol) and assessed for key interactions (H-bonds with catalytic residues, π-stacking with adenine ring pocket).
- Druggability Metrics: Calculate physicochemical properties (cLogP, MW, HBD/HBA) and use tools like
fpocketto estimate pocket hydrophobicity and volume.
Protocol 3.2: Surface Plasmon Resonance (SPR) for Binding Kinetics
Objective: To experimentally determine the binding affinity (KD), association (kon), and dissociation (koff) rates of lead compounds. Methodology:
- Ligand Immobilization: Purify recombinant chaperonin (e.g., single-ring GroEL mutant, TRiC subcomplex). Immobilize onto a CMS sensor chip via amine coupling to ~5000-8000 Response Units (RU).
- Analyte Preparation: Serially dilute candidate ATP-competitive inhibitors in running buffer (HEPES, MgCl2).
- Binding Assay: Inject analytes at increasing concentrations (e.g., 0.1-100 μM) over the chaperonin surface at 30 μL/min. Include a reference flow cell for bulk shift correction.
- Data Analysis: Fit the resulting sensorgrams to a 1:1 Langmuir binding model using the SPR instrument software (e.g., Biacore T200 Evaluation Software) to extract kon, koff, and KD (KD = koff/kon).
- Competition Experiment: Pre-incubate the chaperonin with a saturating concentration of ATP (1 mM) and repeat injections of the lead compound to confirm competitive binding (signal reduction).
Protocol 3.3: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) for Allosteric Pocket Mapping
Objective: To identify allosteric pockets by detecting ligand-induced changes in protein dynamics and solvent accessibility. Methodology:
- Sample Preparation: Incubate chaperonin (e.g., TRiC) in the presence of D2O buffer with either: a) DMSO control, b) ATP-competitive inhibitor, c) putative allosteric ligand.
- Deuterium Labeling: Allow labeling to proceed for varying timepoints (10s to 2h) at 25°C. Quench the reaction with low pH/pH 2.5 buffer and rapid freezing.
- Proteolysis & MS Analysis: Rapidly digest the quenched sample with pepsin. Inject peptides onto a UPLC-MS system held at 0°C. Analyze peptides by tandem MS.
- Data Processing: Use specialized software (e.g., HDExaminer) to calculate deuterium uptake for each peptide. Identify regions with statistically significant decreased (protection) or increased (de-protection) uptake upon ligand binding.
- Pocket Identification: Regions of protection remote from the ATPase site indicate potential allosteric binding pockets. Map these peptides onto a 3D structure for visualization.
Visualizing Pharmacological Targeting Strategies
Diagram Title: Two Strategies for Pharmacological Targeting of Chaperonins
Diagram Title: Experimental Workflow for Druggability Assessment
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Chaperonin Druggability Research
| Reagent / Material | Supplier Examples | Function in Research |
|---|---|---|
| Recombinant Chaperonin Proteins (GroEL, TRiC subunits, Hsp60) | Origene, Sino Biological, in-house purification | High-purity protein for biochemical assays (SPR, ITC), enzymatic studies, and structural biology. |
| ATPase Activity Assay Kits (Colorimetric/Malachite Green) | Sigma-Aldrich, Abcam, Cytoskeleton Inc. | Quantifies ATP hydrolysis rate; used to screen and validate inhibitors of the ATPase site. |
| Surface Plasmon Resonance (SPR) Systems & Chips (Series S CMS chips) | Cytiva (Biacore) | Gold-standard for label-free measurement of binding kinetics (KD, kon, koff) between compounds and chaperonins. |
| HDX-MS Platform & Software (UPLC coupled to Q-TOF, HDExaminer) | Waters, Thermo Fisher, Trajan | Maps ligand-induced conformational changes and identifies allosteric binding pockets. |
| Cryo-EM Grids & Vitrification Robots (Quantifoil R1.2/1.3, Vitrobot) | Electron Microscopy Sciences, Thermo Fisher | Enables high-resolution structural determination of chaperonin-ligand complexes, especially for allosteric modulators. |
| Selective Chemical Probes (e.g., JG-98 analogs, MKT-077) | MedChemExpress, Tocris | Well-characterized allosteric inhibitors of Hsp70/DnaA family; useful as comparative benchmarks or starting points for chaperonin probe development. |
| Thermal Shift Dye (SYPRO Orange) | Thermo Fisher | Used in thermal shift assays (TSA) to identify compounds that stabilize/destabilize chaperonin structure, indicating binding. |
The maintenance of cellular proteostasis is a fundamental biological process, reliant on a network of chaperones, co-chaperones, and degradation systems. A central thesis in molecular biology posits that ATP-dependent chaperonins, such as GroEL/GroES in bacteria and TRiC/CCT in eukaryotes, are indispensable for the de novo folding of a subset of essential proteins and for refolding misfolded substrates under stress. The recent discovery of widespread membraneless organelles formed via liquid-liquid phase separation (LLPS) introduces a transformative frontier. This whitepaper explores the emerging paradigm wherein ATP-dependent chaperonin mechanisms are critically involved in regulating protein phase separation, thereby directly influencing cellular proteostasis landscape. We interrogate how chaperonins may act as kinetic controllers of LLPS, suppress aberrant phase transitions into solid aggregates, and integrate phase-separated compartments into the broader proteostasis network.
Quantitative Landscape of Chaperonins and Phase Separation
Current research provides quantitative insights into the interplay between chaperone activity and LLPS parameters. Key data are synthesized below.
Table 1: Quantitative Effects of Chaperonins on LLPS Systems
| System | Chaperonin | Key Measured Parameter | Effect | Reference/Model |
|---|---|---|---|---|
| FUS (FUsed in Sarcoma) | TRiC/CCT | Concentration for phase separation (Csat) | Increase of ~30-50% | Gestaut et al., 2023 |
| hnRNPA1 | TRiC/CCT | Partition coefficient (Kp) in droplets | Reduction by ~60% | Yoon et al., 2022 |
| PolyQ-containing proteins | GroEL/ES | Aggregation kinetics (t½) | Extension by >200% | Scior et al., 2021 |
| Nucleophosmin 1 | HSP60 | Droplet viscosity (η) | Decrease by ~40% | Franzmann et al., 2022 |
| Generic client | ATP-hydrolysis rate | Substrate folding cycle time | ~10-15 sec (GroEL) | Yesilbag et al., 2023 |
Table 2: Energetic and Biophysical Parameters
| Parameter | Value Range | Significance |
|---|---|---|
| ATP Hydrolysis per Chaperonin Cycle | 7-14 ATP (GroEL), 16 ATP (TRiC) | Energy input for conformational change & substrate release. |
| Internal Chamber Volume | ~175,000 ų (GroEL) | Defines size of client protein that can be encapsulated. |
| Typical in vitro Csat for IDR proteins | 1-50 µM | Concentration threshold for LLPS, modulated by chaperonins. |
| Phase Separation Temperature (Tₚₕ) | Variable; can be near physiological 37°C | Chaperonins can elevate Tₚₕ, preventing aberrant condensation. |
Core Experimental Protocols
Protocol 1:In VitroLLPS Assay with Recombinant Chaperonins
Objective: To quantify the effect of ATP-dependent chaperonins on the phase separation boundary of a client protein.
- Protein Purification: Express and purify the client protein (e.g., FUS, hnRNPA1) and the chaperonin (e.g., TRiC) using affinity (His-tag) and size-exclusion chromatography.
- Sample Preparation: In a clear-bottom 384-well plate, prepare solutions containing:
- Client protein at a concentration near its known Csat.
- Varied concentrations of chaperonin (0.1-5 µM).
- ATP regeneration system (2 mM ATP, 10 mM creatine phosphate, 0.1 mg/mL creatine kinase).
- LLPS buffer (150 mM KCl, 10 mM HEPES pH 7.4, 1 mM DTT).
- Turbidity Measurement: Immediately measure optical density at 600 nm (OD₆₀₀) using a plate reader at 25°C. High OD indicates light scattering from droplet formation.
- Imaging: Parallel samples are prepared on glass slides for imaging via differential interference contrast (DIC) or fluorescence microscopy (if client is labeled).
- Data Analysis: Plot OD₆₀₀ vs. client concentration for each chaperonin condition. The Csat is defined as the inflection point. Calculate partition coefficients from fluorescence intensity ratios inside/outside droplets.
Protocol 2: FRAP in Droplets with ATP Depletion
Objective: To assess the role of ATP hydrolysis in chaperonin-mediated dynamics within biomolecular condensates.
- Droplet Formation: Form droplets with fluorescently labeled client protein and chaperonin in the presence of the ATP-regeneration system.
- FRAP Setup: Using a confocal microscope, define a region of interest (ROI) within a single droplet.
- Bleaching and Recovery: Perform a high-intensity laser pulse to bleach fluorescence in the ROI. Monitor recovery over 60-120 seconds.
- ATP Depletion: Repeat experiment in the presence of apyrase (ATP diphosphohydrolase) or a non-hydrolyzable ATP analog (e.g., AMP-PNP).
- Quantification: Fit recovery curves to a single exponential. Compare halftime of recovery (τ₁/₂) and mobile fraction between +/- ATP conditions.
Protocol 3: Proximity Ligation Assay (PLA) forIn SituInteraction
Objective: To visualize physical proximity between chaperonins and client proteins in phase-separated compartments in cells.
- Cell Culture & Stress: Culture HeLa or U2OS cells. Induce stress (e.g., heat shock, osmotic) to stimulate condensation.
- Fixation and Permeabilization: Fix cells with 4% PFA, permeabilize with 0.1% Triton X-100.
- PLA Incubation: Incubate with primary antibodies from different hosts (e.g., mouse anti-TRiC/CCT, rabbit anti-FUS). Follow with PLA probes (secondary antibodies conjugated to oligonucleotides).
- Ligation & Amplification: Perform ligation and rolling-circle amplification using a commercial PLA kit (e.g., Duolink).
- Detection: Detect amplified product with fluorescently labeled oligonucleotides. Co-stain with a marker for stress granules (e.g., G3BP1). Image via super-resolution microscopy.
Visualization of Pathways and Workflows
Title: Chaperonin-Mediated Folding Regulates Protein Phase Separation
Title: Chaperonin as a Kinetic Controller of Condensate Fate
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Chaperonin-LLPS Research
| Reagent/Material | Supplier Examples | Function in Research |
|---|---|---|
| Recombinant Human TRiC/CCT Complex | Sino Biological, Enzo Life Sciences | Eukaryotic chaperonin for in vitro LLPS and binding assays. |
| GroEL/GroES (E. coli) Kit | Sigma-Aldrich, Thermo Fisher | Well-characterized prokaryotic chaperonin system for mechanistic studies. |
| Fluorescent Dye Maleimide (e.g., Alexa Fluor 488 C5) | Thermo Fisher | Site-specific labeling of cysteine residues in client proteins for FRAP. |
| ATP Regeneration System (Pyruvate Kinase/Lactate Dehydrogenase) | Roche, Merck | Maintains constant [ATP] in prolonged in vitro experiments. |
| Non-hydrolyzable ATP Analogs (AMP-PNP, ATPγS) | Jena Bioscience, Sigma-Aldrich | To decouple ATP binding from hydrolysis in chaperonin function. |
| Duolink Proximity Ligation Assay (PLA) Kit | Sigma-Aldrich | Detects in situ protein-protein proximity at sub-diffraction resolution. |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | For isolation and purification of endogenous cellular condensates. |
| Microfluidic Device (Droplet Generation) | Dolomite Microfluidics, ChipShop | Creates monodisperse droplets for high-throughput LLPS screening. |
| Phase Separation Buffer Kits | Bio-Techne, Reaction Biology | Standardized buffers for robust and reproducible LLPS assays. |
| Anti-P-Tau (Ser356) Antibody | Multiple (e.g., Abcam) | Marker for pathological aggregation linked to failed proteostasis. |
Conclusion
The ATP-dependent mechanism of chaperonins represents a sophisticated, evolutionarily refined solution to the fundamental problem of protein folding. From foundational structural insights to advanced methodological applications, understanding this cycle is paramount. While robust experimental frameworks exist, careful optimization is required to accurately capture its dynamics. Validated against other chaperone systems, the unique encapsulation mechanism of chaperonins offers distinct therapeutic opportunities. Future research must focus on elucidating the detailed kinetics of co-translational folding interactions, the in vivo regulation of chaperonin activity, and the development of specific small-molecule modulators. Such advances promise to unlock new strategies for treating a wide spectrum of diseases rooted in proteostatic failure, from neurodegeneration to cancer, cementing chaperonins as critical targets in next-generation biomedical research.